
Abstract
Outcomes of cardiovascular procedures, such as angioplasty and stent or bypass grafting are limited by failure, predominantly caused by pathological smooth muscle cell (SMC) proliferation, known as intimal hyperplasia. Local delivery of a genetically engineered herpes simplex virus (HSV) is known to block vascular SMC proliferation while allowing for re-endothelialization. However, the mechanism this mutant virus uses to prevent SMC hyperplasia is unknown. The Ras signaling cascade is activated in SMCs undergoing hyperplasia leading to phosphorylation of the mitogen-activated protein kinase (MAPK). In this study we tested the hypothesis that MAPK kinase (MEK) activity is the molecular basis by which SMCs are susceptible to mutant HSV. We show that genetically engineered herpes simplex-1 viruses (HSV-1) can target proliferating SMCs. We demonstrate that the molecular basis of this HSV-1 anti-proliferative effect is MEK activation in SMCs. We demonstrate efficacy and practicality of the MEK-dependent HSV-1 for the treatment of intimal hyperplasia in a clinically relevant in vivo model. Important to this strategy is the ability to modulate the effects by controlling viral dose. These results propel genetically engineered HSV-1 therapy towards clinical evaluation in treatment of intimal hyperplasia.
Similar content being viewed by others


Introduction
Cardiovascular disease is the major cause of mortality in the United States. In 2006, 1 in 2.9 deaths were attributed to cardiovascular disease.1 In all, 1.3 million percutaneous coronary interventions and 448 000 coronary artery bypass procedures and ∼125 000 peripheral bypasses were performed that same year. Up to 29% of coronary bypasses fail at 12–18 months2, 3 and 40% of peripheral bypasses fail at 1 year.4 It is the pathological proliferation of smooth muscle cells (SMCs) of the venous or arterial wall, known as intimal hyperplasia, which leads to operative failure. This SMC proliferation is a form of adaptive remodeling that is due to a change in the local environment. Adaptive remodeling is multifaceted and includes mechanical or hemodynamic injury to the endothelial layer, platelet adhesion and activation, leading to cell proliferation, apoptosis,5, 6 migration, inflammation7 and extracellular matrix production. This process occurs under normal homeostatic conditions as well as pathological conditions, and it is the cellular response to these extracellular stimuli that determines the fine balance between cell survival and death.
The Project of Ex-vivo Vein Graft Engineering via Transfection (PREVENT) trials utilized a genomic approach to alter the balance of cellular proliferation in an attempt to reduce intimal hyperplasia and reduce bypass graft failure. The drug, edifoligide, an antisense oligonucleotide to E2F transcription factor, failed to prove clinical efficacy in the prevention of intimal hyperplasia in coronary or peripheral bypass grafts.2, 4 Speculation as to the reason for failure has focused on the fact that edifoligide is a selective inhibitor for only a subset of the three E2F family members (1, 2 and 3). Thus, cellular redundancy of E2F may have allowed the cell to neutralize the effects of the drug. In addition, dose and efficient transfection may have had contributing roles.8 There have been no further trials for the prevention of bypass graft restenosis. Conversely, drug eluting stents used in the coronary artery circulation, have been successful in preventing intimal hyperplasia,9, 10 however, this success has not translated into the peripheral circulation.
Identifying and characterizing the signaling pathways implicated in SMC proliferation and migration is critical for designing therapeutic strategies targeting intimal hyperplasia. Relevant to this study, the Ras/MAPK kinase (MEK)/mitogen-activated protein kinase (MAPK) cascade has been shown to be an important pro-survival signaling cascade implicated in arterial injury models of intimal hyperplasia.11 Activation of Ras initiates a series of serine/threonine protein kinase phorphorylations including MEK, which then phosphorylates MAPK (ERK1 and ERK2).12, 13 This signaling pathway is known to be activated by alteration of hemodynamic forces such as shear stress and wall tension14 and leads to activation of several pro-survival substrates: c-Fos, c-Jun.15, 16 The prevalence of the Ras/MEK/MAPK pathway in pathological adaptive remodeling, has led to the hypothesis that this is a significant pathway to target for prevention of SMC proliferation.
Recently reported are several genetically engineered, highly attenuated herpes simplex-1 viruses (HSV-1) strains that have been shown to be effective in prevention of vein graft and arterial intimal hyperplasia.17, 18 Of the mutants tested, the more promising are based on deletions of the γ1 34.5 genes. The gene product, infected cell protein (ICP) 34.5 is a multifunctional protein engaged primarily in blocking host defense mechanisms. In particular, double-stranded RNA in host cells will activate double-stranded RNA-dependent protein kinase (PKR), which then phosphorylates eukaryotic translation initiation factor 2α (eIF-2α) preventing it from binding the small ribosomal subunit, blocking the formation of ribosomes and therefore translation. ICP 34.5 acts as a phosphatase accessory factor recruiting protein phosphatase 1α to dephosphorylate the α subunit of eIF-2α.19 As a consequence, protein synthesis continues unimpaired in cells in which the PKR pathway is intact.20, 21, 22 In tumors with constitutive MEK activity, viral activation of PKR is suppressed, thereby rendering cells susceptible to viral replication and cytolysis by γ1 34.5 deleted mutant viruses.23 Mutants lacking the γ1 34.5 gene tested in oncology patients in phase 1 studies have been shown to cause self limiting infections with no virus spread or serious adverse effects.24, 25
The two HSV-1 mutants used in this study were R3616 (Figure 1a), which lacks both copies of the γ1 34.5 gene and R7020, (Figure 1b) which lacks the genes encoding UL23, UL24 and UL56 and the 14.5 Kb internal inverted repeats, which includes one copy of the γ1 34.5 gene. R3616 has a very high margin of safety: intra-cerebral injection of G207 (similar to R3616 but with an additional insertion mutation of ICP 6) at doses as high as 3 × 109 plaque forming units (pfu) into patients with the malignant brain tumor glioblastoma multiforme caused no serious adverse effects.24 In prior reports we showed that R3616 and R7020 are efficacious in the treatment of intimal hyperplasia,17, 18 but the question arose whether engineered HSV-1 virus’ anti-proliferative activity, as it applies to the cardiovascular system, was dependent on activated MEK. As a control we used the R7020 mutant. As its single copy of the γ1 34.5 gene is sufficient without MEK activation to overcome the host defense mechanism of protein synthesis inhibition, it could be predicted that R7020's anti-proliferative activity as it applies to a cardiovascular model of hyperplasia would not require activated MEK.

Schematic representations of R3616 and R7020 HSV-1 genome. (a) Line 1, arrangement of HSV-1 genome (>152 kb). The HSV-1 genome consists of two unique sequences represented as lines, long (UL) and short (US) flanked by inverted repeat sequences ab and b’a’, and a’c’ and ca, respectively, represented as rectangles. Line 2, expansion of the γ134.5 gene loci in the ab and b’a’ inverted repeats. Line 3 shows R3616 virus genome with deleted wild-type DNA locations, where the two γ134.5 genes were located marked by ∣ ∣. (b) Line 1, arrangement of HSV-1 genome (>152 kb). Line 2 shows the expansion of the b’a’ and a’c’ internal repeat and a section of the UL showing the position of the γ134.5 gene in relation to other genes in the repeat. Line 3, schematic representation of the segment deleted HSV-1(F). Line 4 depicts where the internal set of the inverted repeats, containing the genes α0, α4, and γ134.5, was replaced with a construct that contained UL23 fused to the α4 promoter-regulatory region and 3′ to a series of HSV-2 genes. The terminal inverted repeat (ca) remained intact.
Results
MEK activity is required for anti-proliferative activity of R3616
To determine the dose effect of PD98059 and U0126 on MEK activity, triplicate cultures of human aortic SMCs (HAOSMCs) were serum starved overnight followed by exposure to MEK inhibitors PD98059 and U0126 at concentrations ranging from 2 to 20 μM and 2 to 10 μM, respectively, in the absence of serum for 1 h. Serum (10%) was then added to the media to activate MEK and the cells were cultured for an additional 15 min. MEK inhibition as determined by the phosphorylated state of ERK 1/2 was successfully achieved with both inhibitors. U0126 at a dose of 5 μM resulted in near complete inhibition of MEK whereas PD98059 at a dose of 5 μM maintained some MEK activity but it was much less than that in serum starved cells (Figures 2a and b). To investigate the relationships of SMC proliferation and ICP 34.5 function with MEK activity the following experiment was performed. HAOSMCs were cultured overnight in serum free growth medium followed by exposure to U0126 (0–20 μM) for 6 h. The cells were then cultured in M199V with U0126 (0–20 μM) and either R3616 or the ICP 34.5 functionality control virus R7020 at a multiplicity of infection (MOI) of 1, or in the absence of virus for 2 h. After infection, cells were cultured for 72 h in growth medium with 10% serum and U0126 (0–20 μM) and an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed to determine cell survival. The survival of cells, which were treated with U0126 at concentrations >5 μM and then infected with R3616 was statistically equivalent to that of uninfected cells. When MEK was not completely blocked, at U0126 concentrations <5 μM (Figure 2c), cell survival was significantly less than uninfected cells. However, cell survival was significantly decreased for R7020 infected HAOSMCs at all U0126 concentrations. Thus, MEK activity is required for R3616 anti-proliferative activity, but not R7020.

Dose response of ERK 1/2 phosphorylation and viral exposed HAOSMC survival to MEK inhibitors. (a) Western images of phosphorylated ERK, total ERK and GAPDH in HAOSMCs exposed to MEK inhibitors PD98059 and U0126. HAOSMCs serum starved overnight were cultured in serum free medium for 1 h with vehicle (DMSO, lane 1 and 2), with PD98059 (lanes 3–6) and with U0126 (lanes 7, 8 and 9). After which serum was added to the media (lanes 2–9) or the cells remained in serum free medium (lane 1) and cultured for an additional 15 min. (b) Graphed are the western image intensities of phosphorylated ERK 1 and 2 (pERK-1, pERK-2) normalized to GPADH and expressed as percentage of base protein level (lane 1). (c) Survival of HAOSMCs treated with MEK inhibitor U0126 72 h after exposure to R3616 and R7020. Graphed as a percentage of control cells (cultured in the absence of U0126 and virus). Statistically significant difference in cell survival when compared with control cells indicated by * for P<1 × 10−15 and § for P<0.001.
Inhibition of MEK results in an intact PKR pathway
To determine if the target of activated PKR, eIF-2α remains non-phosphorylated and active when MEK activity is induced, despite the presence of HSV, triplicate cultures of HAOSMCs were serum starved overnight and then cultured in the presence and absence of U0126 (5 μM) for 6 h in non-supplemented growth medium followed by a 2 h exposure to R3616 or R7020 at MOIs of 1 in M199V in the presence and absence of U0126. Following infection the cells were cultured in supplemented growth medium with U0126 for 1 and 12 h. In the absence of U0126, MEK activity resulted in very low levels of phosphorylated eIF-2α (p-eIF2α) in cells exposed to either R3616 or R7020 (Figure 3A). When MEK was inhibited with U0126, eIF-2α was phosphorylated in SMCs exposed to the viruses, but to a lesser extent in R7020 infected cells. Quantitative analysis of the images (Figure 3B) confirmed p-eIF2α levels were significantly greater in R3616 infected HAOSMCs in the presence of U0126 than in the absence of the MEK inhibitor (1.9±0.4 vs 7.3±0.6; P<1 × 10−4). Thus MEK inhibition resulted in an intact PKR pathway resulting in phosphorylation of eIF-2α.

Inhibition of MEK allows eIF-2α phosphorylation and inhibits protein synthesis. (A) Western image showing relative levels of p-eIF2α in HAOSMCs. Serum starved cells were cultured in the absence (lanes 1–3) and presence of U0126 (lanes 4–7) for 6 h without serum. After which, they were exposed for 2 h to no virus (control), R3616 (lanes 2, 4 and 5) or R7020 (lanes 3, 6 and 7) under initial MEK inhibitor conditions. This was followed by culturing with serum for 1 h (lanes 1–4 and 6) or 12 h (lanes 5 and 7). (B) Graphed are the western image intensities for p-eIF2α normalized to GAPDH and expressed as a percentage of control. A statistically significant difference in intensity when compared with cells exposed to R3616 without U0126 is indicated by the *P< 1 × 10−4. (C) Western image showing relative levels of late phase viral protein US11 in HAOSMCs. Serum starved cells pre-cultured in the presence (lanes 1, 2, 5 and 6) and absence (lanes 3 and 4) of U0126, were exposed to R3616 (lanes 3–6) or no virus (lanes 1 and 2) for 2 h, and then cultured with serum and U0126 for an additional 12 (lanes 1, 3 and 5) or 24 h (lanes 2, 4 and 6). (D) Western image showing relative US11 expression levels in R7020 infected HAOSMCs cultured without U0126 at 24 h post infection (lane 1) and R7020 exposed cells cultured with U0126 at 24 (lane 2) and 48 (lane 3) h after infection. (E) Immunocytochemistry images of Vero (a–c) and HAOSMCs (d–i) cultured in the absence of virus (a, d and g) and presence of R3616 (b, e and h) or R7020 (c, f and i). HAOSMCs were cultured with U0126 (g, h and i) and without (d, e and f). Cells were stained for US11 (green) and α-actin (red). Nuclei were labeled with DAPI (blue). Note the persistence of US11 in all R7020 images (c, f and i), while in cells infected with R3616 without MEK inhibition it localized to the cytoplasm (e) whereas there was no evidence of US11 with U0126 (h).
To confirm that the phosphorylation of eIF-2α resulted in a shutoff of protein synthesis, western analysis was used to assay for expression of the late phase viral protein US11. Consistent with the hypothesis, MEK inhibition resulted in reduced accumulation of US11 in R3616 (Figure 3C), but not in R7020 (Figure 3D) infected cells. To further confirm that translation was blocked, HAOSMCs exposed to either R3616 or R7020 with and without MEK inhibition were analyzed by immunocytochemistry for US11 expression (Figure 3E). US11 was detected in HAOSMCs without MEK inhibition but, when MEK was inhibited, US11 was only present in cells infected with the R7020 virus, and not R3616. Quantification of immunofluorescence intensity revealed that the presence of U0126 completely abrogated the expression of US11 24 h post R3616 infection (81.4±3.6% in R3616 alone vs 0±0% in R3616+U0126; P<1 × 10−16). Comparatively, R7020 infected cells had a modest yet significant decrease in US11 with the addition of U0126 (140±7% in R7020 alone vs 101%±3% in R7020+U0126; P<1 × 10−4).
Inhibition of MEK in HAOSMCs treated with R3616, resulted in increased levels of p-eIF-2α and a decrease in the late phase viral protein, US11. On the other hand, infection of HAOSMCs with R7020 resulted in accumulation of late phase viral protein US11 despite MEK inhibition. Thus, MEK activation seems to impair the natural host PKR pathway response as shown by the reduction in phosporylated eIF-2α and confers susceptibility to dual deletion γ134.5 mutants.
In vivo MEK activation results in increase viral activity
To confirm that the viral replicative cycle was occurring in vivo, quantitative reverse transcriptase PCR (RT—PCR) was used to assay for HSV-1 US11 gene expression in rabbit carotid arteries exposed to either R3616 (n=3) or R7020 (n=2) in the balloon injury low flow model. Expression levels were also analyzed in arteries that were not operated on (control, n=2) and in arteries that were exposed to R3616 but did not experience balloon injury or low flow (n=3). As seen in Figure 4, the expression of US11 was detected in virus infected arteries indicating viral replication was occurring. Furthermore in injured arteries with low flow, expression levels were greater in those exposed to R7020 than to R3616.

Expression of the gene encoding the late phase protein US11 in vivo. Graph shows mRNA level for the late phase protein US11 normalized to 18s ribosomal RNA quantified by qRT-PCR. The mean cycle threshold for the HSV late phase protein US11 in rabbit carotid arteries exposed to R3616 or R7020 was 21.8 and 20.8, respectively. Total RNA was isolated from carotid arteries that (1) were not operated on (control, n=2), (2) were exposed to R3616 with normal flow and no injury (R3616, n=3), (3) after balloon angioplasty were exposed to R3616 and low flow (R3616+injury, n=3) and (4) were exposed to R7020 after balloon angioplasty with low flow (R7020+injury, n=2).
HSV-1 blocks intimal hyperplasia in a dose-dependent fashion in vitro and in vivo
To test the hypothesis that SMC proliferation is blocked in a dose-dependent fashion, HAOSMCs were exposed to either R3616 or R7020 at concentrations ranging from an MOI of 0 to 5 for 2 h. 72 h after exposure an MTT assay was performed. Both R3616 and R7020 caused a dose-dependent reduction in the number of HAOSMCs (Figure 5a). At high doses, both viruses significantly decreased HAOSMC levels. To determine whether this reduction was mediated directly by the virus, or by the release of autocrine factors, HAOSMCs were cultured in medium conditioned by viral infected cells. Media conditioned for 48 h had no significant anti-proliferative effect compared with non-conditioned growth medium after 24, 36 and 48 h of culture. Thus, the anti-proliferative effect in vitro seems to be mediated in a dose-dependent fashion by the virus directly and not through an autocrine factor (Figure 5b).

Dose response showing decreasing SMC proliferation with increasing viral doses is independent of an autocrine factor. (a) HAOSMC survival 72 h after treatment with R3616 and R7020 at increasing MOIs quantified by MTT assay. Survival was calculated as a percentage of survival for non-infected cells. (b) Survival of HAOSMCs cultured with media conditioned by R3616 and R7020 infected HAOSMCs. Cell survival is expressed as percentage of survival for cells cultured for 24 h with media conditioned by cells cultured in the absence of virus.
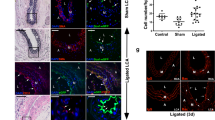
To corroborate the in vitro findings of a dose-dependent response of SMCs to R3616, we determined if there is a dose response to the inhibition of intimal hyperplasia by R3616 in a New Zealand white rabbit intimal hyperplasia model. In this series of experiments, the animals were treated accordingly: group 1 underwent a sham surgery (n=6) and groups 2–4 were subjected to balloon angioplasty and carotid ligation to lower shear stress,6 followed by a 10 min intra-arterial exposure to buffered saline (control, n=7), R3616 (n=11) or R7020 (n=12) given as a single dose escalation ranging from 1 × 103 to 1 × 107 pfu ml−1. Hemodynamic measurements were taken to serve as controls and the experiment terminated after 28 days. The representative cross sections of the 1 × 107 pfu ml−1 dose are shown in Figure 6, and the morphological and hemodynamic measurements are in Table 1. Of note, the hemodynamic assessment revealed no significant difference in either shear stress or mean blood flow in all viral groups compared with control. Assessment of morphology demonstrated that R3616 caused a decrease in intimal hyperplasia, the intima:media ratio and neointimal area at all doses, with significance obtained at 1 × 107pfu ml−1 compared with control. R7020 caused a decrease in intimal hyperplasia as assessed by intima:media ratio and neointimal area, at all doses with significance obtained at 1 × 105pfu ml−1 and 1 × 107pfu ml−1. With respect to the overall adaptive response of the blood vessel to injury and low flow, there was no evidence of increased luminal area in those vessels treated with virus. Using immunohistochemistry, the late phase viral protein US11 was localized to SMCs (Figure 7). These in vivo experiments show that neointimal area and the intima:media ratio are effectively reduced by both R3616 and R7020 in a dose-dependent manner with higher doses being more efficacious and both viruses localizing to SMCs.

Histological evaluation reveals a consistent dose response. Weigart van Geason stained histology sections of New Zealand white rabbit carotid arteries balloon angioplastied treated with: PBS, panels a and e; 1 × 107 pfu ml−1 of R3616 (b and f) and 1 × 107 pfu ml−1 R7020 (c and g) and exposed to low flow for 28 days. Panels d and h depict the sham model. The double ended arrow spans the intimal hyperplasia region and the single ended arrows (white in e, f and h, black in g) indicate the location of the internal elastic lamina. Top panels are × 4 magnification and bottom panels are × 40 magnification.

Immunohistochemical evaluation localizes both R3616 and R7020 to SMCs. Immunohistochemistry of rabbit carotid arteries stained with antibody to the late phase protein US11 and counter stained with Hematoxylin at 72 h post exposure at × 100 magnification. The negative control (a) shows no evidence of US11 compared to R3616 (b) and R7020 (c) both of which show localization of US11 in the medial SMCs (arrow points to representative brown immunoprecipitate).
Discussion
The most relevant finding in this study is that R3616 is a potentially new treatment option for the prevention of pathological SMC proliferation based on activated MEK. The findings that human vascular SMCs are infected by the HSV mutant R3616; that the anti-proliferative capacity is virally mediated; and that anti-proliferation is dependent on MEK activity, are also clinically relevant to the vascular proliferative disorder of intimal hyperplasia.
MEK activation seems to impair the natural host response of the PKR pathway as shown by the reduction in phosporylated eIF-2α and confers susceptibility to dual deletion γ134.5 mutants. Inhibition of MEK suppresses R3616's ability to inhibit proliferating vascular SMCs by allowing phosphorylation of eIF-2α and the prevention of protein synthesis (Figure 8). This finding supports the hypothesis that MEK activity confers to R3616 the ability to replicate and inhibit SMC proliferation. The hypothesis is further supported by the data showing that R7020, which has a single copy of the γ34.5 gene maintained anti-proliferative activity against SMCs in the absence of MEK activity. Furthermore, when we evaluated MEK activity in vivo, we showed that there was an increase in the expression of the late phase viral gene, US11 with a corresponding increase in p-ERK. US11 was chosen because it is a γ2 or a late gene with mRNA expression that is dependent on DNA replication.26 In addition to showing increased US11 mRNA levels with increased MEK activity, we also demonstrated by immunohistochemistry that translation of this late gene is occurring. This data suggests that viral activity is occurring where MEK is active and proliferation is occurring. The relevance of this finding is that MEK activity has been shown to modulate pathological adaptive vascular remodeling in animal models27 as well as in humans.14 The finding of an in vivo anti-proliferative effect brought on by an HSV-1 mutant is consistent with our previous study.18

Schematic representation of mechanism for efficacy of R3616. In quiescent vascular SMCs (a), protein kinase R (PKR) mediates an innate host cellular defense against viral infection. When HSV infects a cell, PKR is autophosphorylated, thereby becoming active. PKR then goes on to phosphorylate eIF-2α or eukaryotic initiation factor 2α. eIF-2α is essential for translation, and it's phosphorylation shuts down cellular translation preventing viral replication. Wild-type HSV is able to evade host cellular defenses, in part γ1 34.5 gene product, ICP 34.5 (or infected cell protein 34.5). ICP 34.5 recruits a phosphatase (PP1α) to dephosphorylate eIF-2α returning it to its active state and allowing viral gene translation to proceed. (b) γ1 34.5 mutants such as R3616 lack ICP 34.5 and therefore are unable to overcome this host cellular defense mechanism mediated by PKR and therefore unable to replicate in normal tissues. (c) In SMCs that have been exposed to injury and low shear stress, the Ras/Raf/MEK pathway is activated. MEK suppresses the host antiviral defense of PKR activation. Thus viral replication proceeds even for viral mutants lacking ICP 34.5 expression. MEK expression confers susceptibility to R3616.
Traditionally, strategies to deliver a protein, alter the genome or modulate immunity have been utilized to reduce intimal hyperplasia. For example, in a study designed to evaluate the role of Survivin in vascular lesion formation, local delivery of a replication-deficient adenovirus was used to deliver a dominant-negative Survivin mutant. The virus was used to deliver the phosphorylation-defective Survivin mutant, which resulted in a significant reduction in neointimal mass.28 Another strategy to target intimal hyperplasia that made it to phase III clinical trials utilized an antisense oligonucleotide delivered in an ex-vivo manner directly to the vascular tissue at the time of surgery. The antisense oligonucleotide targeted E2F transcription factor resulting in a block of cell cycle gene expression.2, 4, 29 Unfortunately, these strategies have failed when applied to peripheral arterial disease. The drug eluting stent for the treatment of intimal hyperplasia in the coronary arteries has succeeded9 with important clinical limitation of late stent thrombosis.30 However, this success has not translated into the peripheral circulation. The cytoreductive strategy presented in our studies represents a new and divergent approach from these classical approaches to intimal hyperplasia: the agent itself is anti-proliferative and does not rely on delivering a protein or altering gene expression.
Potential clinical applications for this therapy include utilizing a model in which a vein is harvested at the time of bypass surgery and then perfused with either R3616 or R7020 ex vivo before re-implantation as the bypass graft. This is a clinically applicable methodology previously used in human trials.2, 4 Furthermore, we have shown that we are able to modulate intimal hyperplasia in a relevant vein graft model.17, 31 Similarly, both viruses could be used to coat a balloon or stent or be directly delivered to site of injury in the vessel to prevent intimal hyperplasia at the time of angioplasty and stenting. Furthermore, it might be advantageous to expose the vein to an agent at the time of arterial-venous fistula creation to prevent intimal hyperplasia in dialysis access patients. The clinical use of these viruses is compelling because there is a dose-dependent reduction in intimal hyperplasia in a validated in vivo rabbit model. Importantly, there was no evidence of aneurismal dilatation of treated vessels. This is germane because it is felt that some adaptive remodeling is important to allow for appropriate vascular adaptation but not pathological hyperplasia. The finding of dose-dependence is appealing in the clinical setting by allowing dose adjustments at the time of surgery depending on the risk factors for intimal hyperplasia associated with the patient and procedure.
Despite numerous advances in surgical technique and drug design, there remains no good therapeutic approach to the treatment of intimal hyperplasia in peripheral arteries or vein grafts. In conclusion, we showed that R3616, an engineered HSV-1 with a dual deletion of the γ34.5 gene, resulted in a significant anti-proliferative effect on SMCs based on intrinsic MEK activity; whereas, R7020 with a single deletion of γ34.5 gene acted independent of MEK in SMCs. These data would suggest that current HSV-1 technology is able to effect significant cellular change by reducing MEK-mediated pathological smooth muscle proliferation.
Materials and methods
Cells and viruses
Primary human aortic SMCs were purchased from (Cell Application, Inc., Cat# 354-05a, San Diego, CA, USA) and maintained in SMC growth medium (Cell Application, Inc., Cat#311-500) supplemented with 10% fetal bovine serum. Experiments were performed with cells at passages 3–6. Viral strain preparation has been described elsewhere.32, 33, 34, 35 Vero cells were maintained in DMC supplemented with 5% newborn calf serum.
Dose effect of U0126 and PD98059 on MEK activity
To determine the dose effect of MEK inhibitors U0126 and PD98059 on MEK activity in HAOSMCs, cells were seeded in T-25 flask at a density of 5 × 104 cells cm−2 in supplemented growth medium and cultured at 37 °C with 5% CO2. At 50% confluence they were starved overnight in serum-free growth medium and then exposed to U0126 (Promega, Cat# V1121, Madison, WI, USA) or PD98059 (Promega, Cat# V1191) at concentrations that ranged from 0 to 20 μM and 0 to 10 μM, respectively, in non-supplemented growth medium for 1 h. For cultures with no inhibitor, DMSO (1:1000) was added as a vehicle control. Serum (10%) was then added to the media and the cells were cultured for an additional 15 min before lysis with RIPA Lysis Buffer (Santa Cruz Biotechnology, Cat# sc-24948, Santa Cruz, CA, USA). MEK activity was assessed by western analysis of phosphorylated and non-phosphorylated ERK1/2.
Effect of R3616 and R7020 in conjunction with MEK inhibition on cell survival
To determine the effects of R3616 and R7020 on cell survival in the presence of MEK inhibitor U0126, HAOSMCs were seeded in 96-well plates at a density of 5 × 103 cells per well in 0.1 ml of supplemented growth medium. At 50% confluence they were starved overnight in serum-free growth medium and then exposed to U0126 at concentrations that ranged from 0 to 20 μM in non-supplemented growth medium for 6 h. The HAOSMCs were then cultured in the absence and presence of these viruses at an MOI of 1 in M199V with U0126 (0–20 μM) for 2 h. After the 2 h incubation the culture medium was replaced with 10% fetal bovine serum supplemented growth medium containing U0126 (0–20 μM) and the cells were cultured for 3 days. HAOSMC survival was then quantified with an MTT assay as previously described.31
Culture of HAOSMC for PKR pathway activity in the presence of R3616 and R7020 with MEK inhibition
HAOSMCs were seeded in T-25 flask at a density of 5 × 104 cells cm−2 in supplemented growth medium. At 50% confluence they were starved overnight in serum-free growth medium and then cultured in non-supplemented growth medium in the absence and presence of U0126 (5 μM) for 6 h. After which the medium was replaced with M199V containing R3616 or R7020 at an MOI of 1, or no virus, with and without U0126 (5 μM) for 2 h. The cells were then cultured in serum (10%) supplemented growth medium in the presence and absence of U0126 (5 μM). For analysis of p-eIF-2α, cells were lysed after 1 and 12 h. For analysis of US11 protein levels, cells treated with R3616 were lysed after 12 and 24 h and cells exposed to R7020 after 24 and 48 h.
Western analysis
Total protein concentrations were quantified with a Bradford assay. Protein samples (30–40 μg) were electrophoresed in 12% denaturing polyacrylamide gels (Invitrogen, Cat# NP0341BOX, Grand Island, NY, USA) at 100V for 90 min. After transfer to PVDF membranes (Invitrogen, Cat# LC2005) at 30 V for 90 min, membranes were blocked in Superblock T20 (TBS) blocking buffer (Thermo Scientific, Cat # 37536, Rockford, IL, USA) for 1 h at room temperature (RT). The membranes were then incubated with the appropriate primary antibody for 1 h at RT or overnight at 4 °C. This was followed by three washes for 5 min in TBS with 0.05% TWEEN 20 and incubation with the appropriate secondary antibody conjugated to horseradish peroxidase for 1 h at RT. The membrane was again washed as described above and the secondary antibody was detected with enhanced chemiluminescence using SuperSignal West Femto (Thermo Scientific, Cat# 34095). The membranes were imaged using KODAK Image Station 440cf System (Rochester, NY, USA). Densitometry analysis was performed with NIH ImageJ software (NIH, Bethesda, MD, USA). For quantization, protein levels were normalized to GAPDH. The following antibodies and dilutions were used: polyclonal rabbit anti-human phosphorylated ERK1/2 (1:5000, Promega, Cat# V8031); polyclonal goat anti-human p-eIF-2α (1:200, Santa Cruz Biotechnology, Cat# sc-12412); monoclonal mouse antibody to US1136 was a gift from Dr Roizman; monoclonal mouse anti-human GAPDH (1:200, Santa Cruz Biotechnology, sc-47724); goat anti-rabbit IgG (1:3000, Thermo Scientific, Cat# 1858415) and donkey anti-goat IgG (1:5000, Santa Cruz Biotechnology, Cat# 2020).
Immunocytochemistry
HAOSMCs were seeded on four-well slides at >90% confluence and cultured overnight in supplemented growth medium, they were then cultured overnight in serum-free growth medium. After which the HAOSMCs were exposed to U0126 (5 μM) or DMSO (1:1000, vehicle) for 6 h in non-supplemented growth medium. They were next cultured in M199 V with R3616 or R7020, at an MOI of 1 in the absence and presence of U0126 (5 μM). After 2 h, the media was replaced with supplemented growth medium with and without U0126 (5 μM) for 24 h. Vero cells were seeded on four-well slides at >90% confluence and cultured overnight in DMC supplemented with 5% newborn calf serum after which they were cultured in the presence of R3616 and R2070 or in the absence of virus for 2 h in M199V. They were then cultured for 24 h in supplemented DMC in the absence of virus for 24 h. The cells were fixed in 4% paraformaldehyde in phosphate-buffered solution (PBS) for 15 min at RT, washed with PBS and permeabilized for 40 min at RT in PBS with 10% human serum, 1% bovine serum albumin and 0.1% Triton X-100. The cells were incubated with a mouse antibody to US11 (gift from Dr Roizman) at 4 °C overnight, washed three times for 5 min in TBS with 0.05% TWEEN 20, incubated with a FITC conjugated-Goat anti-Mouse IgG antibody for 1 h at RT (1:64, Sigma, Cat# F2012, St Louis, MO, USA), washed three times for 5 min in TBS with 0.05% TWEEN 20 and then covered in VECTASHIELD mounting medium with DAPI (Vector Laboratories, Cat# H-1200, Burlingame, CA, USA). Images were acquired using an Olympus U-TB190 DSU Spinning Disk Confocal Microscope. Image analysis was performed using NIH ImageJ software (NIH) as previously described18 and outlined in the ImageJ User guide.37 The images presented are representative of the images used for quantification. Quantification of the relative intensity was performed by using Image J software to measure the intensity of 10 high power ( × 600) fields per sample. The threshold levels were based on the positive control (Vero cells) and percentages based on quantification standardized to the positive control.
Animal operations
The animal model utilized has been previously characterized in both rats5 and rabbits.38 Male New Zealand white rabbits (3 kg) were cared for in accordance with the University of Chicago Institutional Animal Care and Use Committee.
The model used was a low flow and balloon injury model of the common carotid artery using the New Zealand white rabbit to induce MEK. Details of the operations were performed as previously described.6, 18 For the dose response experiments, four experimental groups of animals were used with direct intra-arterial delivery of agents for 10 min at a pressure equal to the last measured systolic blood pressure. The control group was treated with 1–2 cc of phosphate buffered saline (control group, n=7); the R3616 group was treated with R3616 at doses of 1 × 103(n=4), 1 × 105 (n=3), 1 × 107(n=4) pfu ml−1; the R7020 group was treated with doses of 1 × 103(n=5), 1 × 105 (n=4), 1 × 107(n=3) pfu ml−1. For the sham group (n=6), the left common carotid artery was temporally exposed as for the other groups and the skin sutured closed as the others. For the quantitative RT–PCR experiments and measurement of p-ERK1/2, four experimental groups were used: a no surgery negative control group (n=2); intra luminal exposure to R3616 (1 × 107 pfu ml−1, n=3) without low flow and balloon injury; low flow and balloon injury with intra luminal exposure to R3616 (1 × 107 pfu ml−1, n=3); low flow and balloon injury with intra luminal exposure to R7020 (n=2).
After 3 days for the PCR experiments and 28 days for the dose response, animals were re-anesthesized and the injured common carotid artery segments was re-exposed. Repeat flow and pressure measurements were made. Four centimeter segments of injured artery were harvested from each rabbit and the animals were euthanized using 120 mg kg−1 IV pentobarbital. For histomorphometric analysis, the artery was perfusion fixed with 1.25% formaldehyde at the last recorded systemic blood pressure for 10 min. For RNA analysis, the arteries were snap frozen immediately in liquid nitrogen.
RNA isolation and RT–PCR
Total RNA was isolated was from rabbit carotid artery using RNeasy Fibrous Tissue Kit (Qiagen, Cat# 74704, Germantown, MD, USA). For each vessel, 0.5 μg of total RNA was reverse transcribed to generate cDNA (Invitrogen SuperScript III First-Strand Synthesis System for RT–PCR, Cat#18080-051). Quantification of gene expression was performed by quantitative PCR (ABI StepOnePlus) using the Invitrogen SYBR GreenER Supermix Universal (Cat#11762-100), which uses the fluorescein compound SYBR-Green for amplicon detection. The primers for US11 were designed to amplify a region in the US11 cDNA that does not overlap with the US10 cDNA. The forward US11 primer was 5′-GGGCGACCCAGATGTTTACTT-3′ and the reverse primer was 5′-TGTCCTCGAGGTGCGTGAA-3′. An 18s primer mix (QuantumRNA Universal 18s Internal Standard, Applied Biosystems, Cat#AM1718M, Carlsbad, CA, USA) was used to amplify 18s ribosomal RNA as an internal control for normalization. PCR conditions consisted of an initial 10 min denaturation step at 95 °C and a 40-cycle amplification program (95 °C, 30 s; 60 °C, 60 s). Threshold cycle readings for each experiment were determined using the same threshold setting, respectively. All reactions were performed in triplicate and each experiment was repeated at least once to ensure consistency.
Immunohistochemistry
Frozen sections were cut in 5–7 μm thick sections. The slides were fixed in ice cold acetone for 6–10 min and washed in PBS with 0.05% TWEEN 20. Slides were subsequently incubated in Peroxidase Block Solution (Dako, Envision+ System-HRP (DAB) Kit, Cat# K4007, Carpinteria, CA, USA) at RT for 5 min, washed two times in PBS with 0.05% TWEEN 20 for 5 min, blocked in PBS or TBS with 3% BSA and 0.05% TWEEN 20 at RT for 30 min, and incubated with a mouse antibody to US11, (1:1000 gift from Dr Roizman) in PBS or TBS with 3% BSA and 0.05% TWEEN 20 at RT for 1 h and then washed in PBS with 0.05% TWEEN 20. The slide was incubated with labeled polymer-HRP anti-mouse IgG antibody (Dako Envision+ System-HRP (DAB) Kit, Cat# K4007) at RT for 30 min and washed in PBS with 0.05% TWEEN 20. DAB+Substrate-Chromogen solution was applied to cover the specimen and incubated at RT for 2–10 min. The samples were counterstained with Hematoxylin.
Viral in vitro dose-response assay
HAOSMCs were seeded in 96-well plates at a density of 5 × 103 cells per well in 0.1 ml of supplemented growth medium and cultured overnight. They were then exposed to R3616 and R7020 at an MOI that ranged from 0 to 5 in M199V. After 2 h, the medium was replaced with supplemented growth medium and cultured for 72 h. HAOSMC viability was quantified with an MTT assay as previously described.31 To determine if autocrine factors were responsible for the dose effect experiments were performed with conditioned media. Conditioned medium was prepared by first exposing HAOSMCs at >95% confluence in a T-25 to R3616 or R7020 at an MOI of 1 in M199V for 2 h. The media was then replaced with growth medium supplemented with 10% fetal bovine serum (5 ml) and cultured for 48 h. To remove viral particles the conditioned media was centrifuged at 40 000 r.p.m. for 1 h after which the supernatant was twice passed through a 0.1 μm filter. HAOSMCs were seeded in 96-well plates at a density of 5 × 103 cells per well in 0.1 ml of supplemented growth medium and cultured overnight. The medium was changed to the conditioned medium (0.1 ml per well) and the cells were cultured for 24, 36 and 48 h. HAOSMC viability was then quantified with an MTT assay as previously described.31
Assessment of morphology
Image analysis for histomorphometry was performed as previously described.18 Intimal thickness, medial thickness, luminal area and medial area were digitally measured using NIH ImageJ software, whereas the intima:media ratio and neointimal area were calculated using the measured values. Shear stress was calculated as 32 μQ/πd3, where μ is the viscosity of blood (0.035 poise); Q is the mean blood volume flow rate (cm3 s−1) and d is the measured diameter.38
Statistical data analysis
Statistical data analysis was performed using standard software (Microsoft Excel Data Analysis; Redmond, WA, USA). Data is presented as mean±s.e.m. Comparison of means was achieved using Student's t-test and one-way analysis of variance where appropriate with a significance assigned at P<0.05. Quantitative RT-PCR data were compared using analysis of variance and unpaired t-tests.
References
Lloyd-Jones D, Adams R, Brown T, Carnethon M, Dai S, De Simone G et al. Heart Disease and Stroke Statistics 2010 Update: a report from the American Heart Association. Circ 2010; 121: e46–e215.
PREVENT Investigators. Efficacy and safety of edifoligide, an E2F transcription factor decoy, for prevention of vein graft failure following coronary artery bypass graft surgery: PREVENT IV: a randomized controlled trial. JAMA 2005; 294: 2446–2454.
Goldman S, Zadina K, Moritz T, Ovitt T, Sethi G, Copeland JG et al. Long-term patency of saphenous vein and left internal mammary artery grafts after coronary artery bypass surgery: Results from a Department of Veterans Affairs Cooperative Study. J Am Coll Cardiol 2004; 44: 2149–2156.
Conte M, Bandyk D, Clowes A, Moneta GL, Seely L, Lorenz TJ et al. Results of PREVENT III: a multicenter, randomized trial of edifoligide for the prevention of vein graft failure in lower extremity bypass surgery. J Vasc Surg 2006; 43: 742–751.
Clowes A, Reidy M, Clowes M . Mechanisms of stenosis after arterial injury. Lab Invest 1983; 49: 208–215.
Spiguel L, Chandiwal A, Vosicky J, Weichselbaum RR, Skelly CL . Concomitant proliferation and caspase-3 mediated apoptosis in response to low shear stress and balloon injury. J Surg Res 2010; 16: 146–155.
Hanke H, Hassenstein S, Ulmer A, Kamenz J, Oberhoff M, Haase KK et al. Accumulation of macrophages in the arterial vessel wall following experimental balloon angioplasty. Eur Heart J 1994; 15: 691–698.
Conti V, Hunter G . Gene therapy and vein graft patency in coronary artery bypass graft surgery. JAMA 2005; 294: 2495–2497.
Suzuki T, Kopia G, Hayashi S, Bailey LR, Llanos G, Wilensky R et al. Stent-based delivery of sirolimus reduces neointimal formation in a porcine coronary model. Circulation 2001; 104: 1188–1193.
Rensing BJ, Vos J, Smits PC, Foley DP, van den Brand MJ, van der Giessen WJ et al. Coronary restenosis elimination with a sirolimus eluting stent: first European human experience with 6-month angiographic and intravascular ultrasonic follow-up. Eur Heart J 2001; 22: 2125–2130.
Hu Y, Cheng L, Hochleitner B, Xu Q . Activation of mitogen activated protein kinases (ERK/JNK) and AP-1 transcription factor in rat carotid arteries after balloon injury. ATVB 1997; 17: 2808–2816.
Seger R, Krebs E . The MAPK signaling cascade. FASEB 1995; 9: 726–735.
Blumer K, Johnson G . Diversity in function and regulation of MAP kinase pathways. Trends Biochem Sci 1994; 19: 236–240.
Curi M, Skelly C, Meyerson S, Baldwin ZK, Lee E, Lanahan J et al. Differential activation of MAPK signal transduction in intact human artery and vein. J Surg Res 2002; 108: 198–202.
Marais R, Wynne J, Treisman R . The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 1993; 73: 381–393.
Zinck R, Hipskind R, Pingoud V, Nordheim A . c-fos transcriptional activation and repression correlate temporally with phosphorylation status of TCF. EMBO J 1993; 12: 2377–2387.
Skelly C, Curi M, Meyerson S, Woo DH, Hari D, Vosicky JE et al. Prevention of restenosis by a herpes simplex virus mutant capable of controlled long-term expression in vascular tissue in vivo. Gene Therapy 2001; 8: 1840–1846.
Skelly C, Chandiwal A, Vosicky J, Weichselbaum RR, Roizman B . Attenuated herpes simplex virus 1 blocks arterial apoptosis and intimal hyperplasia induced by balloon angioplasty and reduced blood flow. Proc Natl Acad Sci 2007; 104: 12474–12478.
He B, Gross M, Roizman B . The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci 1997; 94: 843–848.
Chou J, Chen J, Gross M, Roizman B . Association of a M(r) 90 000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc Natl Acad Sci 1995; 92: 10516–10520.
Cheng G, Brett M, He B . Val193 and Phe195 of the gamma 1 34.5 protein of herpes simplex virus 1 are required for viral resistance to interferon-alpha/beta. Virology 2001; 290: 115–120.
Leib D, Machalek M, Williams B, Silverman RH, Virgin HW . Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc Natl Acad Sci 2000; 97: 6097–6101.
Smith K, Mezhir J, Bickenbach K, Veerapong J, Charron J, Posner MC et al. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Deltagamma(1)34.5 mutants of HSV-1. J Virol 2006; 80: 1110–1120.
Markert J, Medlock M, Rabkin S, Gillespie GY, Todo T, Hunter WD et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase 1 trial. Gene Therapy 2000; 7: 867–874.
Fong Y, Kim T, Bhargava A, Schwartz L, Brown K, Brody L et al. A herpes oncolytic virus can be delivered via the vasculature to produce biologic changes in human colorectala cancer. Mol Ther 2009; 17: 389–394.
Chou J, Roizman B . The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci USA 1992; 89: 3266–3270.
Saunders P, Pintucci G, Bizekis C, Sharony R, Hyman KM, Saponara F et al. Vein graft arterialization causes differenctial activation of mitogen-activated protein kinases. J Thor and Cardiovasc Surg 2004; 127: 1276–1284.
Simosa H, Wang G, Sui X, Peterson T, Narra V, Altieri DC et al. Survivin expression is up-regulated in vascular injury and identifies a distinct cellular phenotype. J Vasc Surg 2005; 41: 682–690.
Mann M, Whittemore A, Donaldson M, Belkin M, Conte MS, Polak JF et al. Ex-vivo gene therapy of human vascular bypass grafts with E2F decoy: the PREVENT single-centre, randomised controlled trial. Lancet 1999; 354: 1493–1498.
Pfisterer M, Brunner-La Rocca HP, Buser PT, Rickenbacher P, Hunziker P, Mueller C et al. Late clinical events after clopidogrel discontinuation may limit the benefit of drug-eluting stents: an observational study of drug-eluting versus bare-metal stents. J Am Coll Cardiol 2006; 48: 2584–2591.
Curi M, Skelly C, Meyerson S, Baldwin ZK, Balasubramanian V, Advani SJ et al. Sustained inhibition of experimental neointimal hyperplasia using a genetically modified herpes simplex virus. J Vasc Surg 2003; 37: 1294–1300.
Meigner B, Longnecker R, Roizman B . In vivo behavior of genetically engineered Herpes simplex viruses R7017 and R7020: Construction and evaluation in rodents. J Infect Dis 1988; 158: 602–614.
Meigner B, Longnecker R, Roizman B . In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020. J Infect Dis 1990; 162: 313–321.
Chou J, Kern E, Whitley R, Roizman B . Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990; 250: 1262–1266.
Andreansky S, Soroceanu L, Flotte E, Chou J, Markert JM, Gillespie GY et al. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res 1997; 57: 1502–1509.
Roller R, Roizman B . The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J Virol 1992; 66: 3624–3632.
Ferreira T, Rasband W . The ImageJ User Guide--IJ 1.45. 2010–2011, (cited 2010); available from: imagej.nih.gov/ij/docs/guide.
Meyerson S, Skelly C, Curi M, Shakur UM, Vosicky JE, Glagov S et al. The effects of extremely low shear stress on cellular proliferation and neointimal thickening in the failing bypass graft. J Vasc Surg 2001; 34: 90–97.
Acknowledgements
We thank Dr Bernard Roizman (University of Chicago) for kindly providing reagents and his expertise. Dr Skelly is funded by NIH K-08-HL091053 as well by the American Vascular Association/American College of Surgeons and NHLBI Jointly Sponsored Mentored Clinical Scientist Development Award. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI or the NIH
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Drs Skelly and Weichselbaum are co-founders of Maji Therapeutics with founders stock.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Skelly, C., He, Q., Spiguel, L. et al. Modulating vascular intimal hyperplasia using HSV-1 mutant requires activated MEK. Gene Ther 20, 215–224 (2013). https://doi.org/10.1038/gt.2012.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gt.2012.26
