
Abstract
Purpose: Recurrence of lethal osteogenesis imperfecta in families results from either dominant (parental mosaicism) or recessive inheritance. The proportion of these two mechanisms is not known, and determination of the contribution of each is important to structure genetic counseling for these families.
Methods: We measured the recurrence rate of lethal osteogenesis imperfecta after the birth of an affected infant. We determined the rate of parental mosaicism in a subset of families in which we had identified dominant mutations. In 37 families in which two or more affected infants were born, we identified mutations and determined the proportion that resulted from recessive inheritance.
Results: The recurrence rate after the first affected pregnancy was 1.3%. The rate of parental mosaicism in families in which a dominant mutation was identified in a first affected child was 16%. In 37 families with two affected infants, 26 had dominant mutations, seven had recessive mutations, and we failed to find mutations in four. The overall recurrence rate for couples after two or more affected infants was 32%; 27% for families with parental mosaicism, 31% for recessive mutations, and 50% for families with no identified mutation.
Conclusions: In most populations, recurrence of lethal osteogenesis imperfecta usually results from parental mosaicism for dominant mutations, but the carrier frequency of recessive forms of osteogenesis imperfecta will alter that proportion. Mutation identification is an important tool to assess risk and facilitate prenatal or preimplantation diagnosis.
Similar content being viewed by others


Main
In the introduction of the modern classification of osteogenesis imperfecta (OI), OMIM# 166200, Sillence et al.1 asserted that the perinatal lethal form of OI (OI type II), OMIM# 166210, was a recessively inherited disorder, based on observed consanguinity and recurrence with a segregation ratio of close to 25%. Subsequent studies from several groups observed that the recurrence risk was usually less than 10%,2 and molecular genetic studies identified heterozygous mutations in type I collagen genes.3–6 Notably, recurrence occurred in some instances in which one parent had affected children with two partners, an observation difficult to explain by recessive inheritance. Studies on the molecular basis of OI in several families then showed that recurrence of OI type II could result from parental mosaicism for dominant mutations in type I collagen genes.7,8
The initial estimates of recurrence9,10 usually resulted from analysis of retrospective data in which ascertainment was probably biased toward families with recurrence. The measured rate of recurrence of OI type II in an unbiased population was 2% after the birth of an affected infant born to unaffected parents.11 These observations did not, however, identify the cause of recurrence in consanguineous families.
In 2006, the second phase of this story began to unfold with the identification of biallelic mutations in CRTAP, OMIM# 610854,12–15 closely followed by the recognition of recessive mutations in genes that encode proteins that interact with CRTAP: LEPRE1, OMIM# 61091515–17 and PPIB, OMIM# 259440 (unpublished data).18,19 In an unselected set of 63 infants with lethal OI, 60 (95%) resulted from mutations in type I collagen genes, with the remainder in the then recognized recessive genes.20
The birth of a second affected child places a family into one of two groups—one in which a parent is mosaic for a dominant mutation or a group in which each parent is a carrier of a recessive form of OI. The calculated recurrence risks for these two groups differ—as high as 50% for families in which one parent is mosaic (depending on the proportion of germ cells that carry the mutation) and 25% for families in which each parent is a carrier. Because the maximum recurrence risk is different in the two circumstances and because approaches to diagnosis differ, it is important to determine the proportion of families that can be attributed to each group and how mutation identification can clarify the risk of recurrence in each family and facilitate prenatal counseling, prenatal diagnosis, or preimplantation diagnosis.
METHODS
Subject selection
As a follow-up to the study by Pepin et al.,11 we identified all prenatal samples submitted to the Collagen Diagnostic Laboratory (CDL) at the University of Washington from June 1996 to August 2009 and measured the recurrences after the identification of lethal OI in one affected infant or pregnancy, and after two or more affected pregnancies. We made the diagnosis of OI type II if the infant died within a month of delivery, if the radiologic picture was consistent with the diagnosis, if the referring physician had made the clinical diagnosis, if the ultrasound during pregnancy was described as consistent with OI type II, or if a mutation was identified that had previously been found in an infant with this condition.
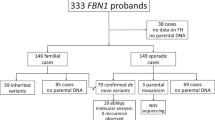
Second, we identified 216 infants (or pregnancies) in the CDL Repository with lethal OI, in which a type I collagen mutation had been found. We had DNA available from 37 parental pairs of the 216 affected infants to determine whether a parent was mosaic for the mutation.
Finally, to measure the proportion of pregnancies that could be attributed to dominant or recessive mutations, we identified a separate set of 37 families in which there were two or more affected pregnancies born to unaffected parents from the CDL Repository (approved by the Institutional Review Board of the University of Washington). The sequences of the coding and splice site regions of COL1A1, COL1A2, CRTAP, LEPRE1, and PPIB were determined to identify the causative mutations.
Analysis of candidate genes
RNA or genomic DNA was extracted from cultured dermal fibroblasts using the QIAamp® RNA or DNA Mini Kit (Qiagen, Chatsworth, CA), and cDNA was synthesized using the SuperScript® First Strand kit (Invitrogen, San Diego, CA). The coding sequences and flanking intron regions of COL1A1 and COL1A2 were amplified in 17 and 23 fragments, respectively, and sequenced in multiple reactions. The coding sequences of CRTAP and LEPRE1 were amplified and sequenced as described.15 The coding sequences and flanking intron regions of PPIB were amplified and sequenced using five products. The sequence traces were analyzed with Mutation Surveyor® software. The primers and reaction conditions are available on request from the authors.
Determination of recurrence risk
The recurrence rate in all classes of sibships was calculated by excluding the first affected infant(s) in the sibship and then counting the subsequent affected and unaffected infants in subsequent births.
RESULTS
Recurrence rate of lethal OI among prenatal diagnosis samples
Between June 1996 and August 2009, 76 families, in which there was one previously affected pregnancy, requested prenatal diagnosis studies. In each instance, either biochemical analysis (60 families) or genetic analysis (16 families, Table 1) had confirmed the clinical diagnosis of OI type II in the first affected pregnancy. In the 76 pregnancies we studied, there was one recurrence (1/76, 1.3%) (Table 2). Seven families in which there were two previously affected pregnancies requested prenatal diagnosis, and in those, there were two recurrences (2/7, recurrence rate of 29%). Finally, for three families in which there were three previously affected, two of the three pregnancies were affected (67% recurrence rate). These rates are similar to those previously identified.11
Parental mosaicism for dominant mutations
We identified mutations in type I collagen genes (COL1A1 or COL1A2) in 216 index patients with OI type II. In 37 of these families, DNA from blood or fibroblasts derived from the parents was sequenced around the site of the mutation identified in the affected child to determine whether the causative mutation was present in a subset of alleles in one parent. In 6 of the 37 families (16%), one parent was found to be mosaic (four mothers and two fathers). Of those six, two (both mothers) were noted to have clinical features of mild OI.
Proportion of dominant compared with recessive inheritance after recurrence
By searching our Repository, we identified an additional 37 families in which two or more affected infants had been born to unaffected parents. We sequenced five candidate genes, COL1A1, COL1A2, CRTAP, LEPRE1, and PPIB, to identify causative mutations (Table 3). Cultured fibroblasts from all but one of these infants produced some abnormal type I collagen molecules in which the constituent chains were overmodified and migrated slowly during gel electrophoresis. In 26 of the 37 (70%) families (67 children with 56 affected), we found mutations in a type I collagen gene, COL1A1 (15) or COL1A2 (11) (Table 3). In 7 of the 37 (19%) families (27 children with 18 affected), we found homozygosity or compound heterozygosity for mutations in CRTAP (3), LEPRE1 (3), or PPIB (1). We failed to find mutations in the remaining four (11%) families (14 children with 11 affected) (Table 3).
Determination of recurrence risk
At the time we ascertained the 37 families in which there were two or more children with OI type II, there had been 108 pregnancies, of which 85 were affected (Table 4). After accounting for ascertainment, the overall rate of recurrence in these families was 32% (11/34). In families with parental mosaicism for dominant mutations, the recurrence rate was 27% (4/15); in families with recessive mutations, the recurrence rate was 31% (4/13); and in families with no mutation identified, the recurrence rate was 50% (3/6). Because the number of families is small, the measured rates serve as estimates that could change if sample size is increased.
Mosaicism among parents with recurrent OI due to dominant mutations
In 26 families, in which two or more affected infants were born, a type I collagen mutation was identified in an affected child. We studied parental blood samples from 16 of those families to determine the proportion in which we could identify the mutation found in the child and whether there was a higher rate of mosaicism in one gender. Among those 16, we identified the mosaic parent in 15 (seven mothers and eight fathers) families. Two of the parents were assigned as mosaic on the basis of family structure (i.e., they had affected children with different partners—one mother and one father). The status was determined by DNA analysis in the remaining 13 among whom we identified mutations in 12. Among the 15 parents determined to be mosaic, three had signs compatible with mild OI (one of the fathers8 and two mothers). The extent of mosaicism in blood and fibroblast DNA varied, but in only one family did the proportion of affected alleles approach 50% of the total in blood DNA. The proportion of affected alleles in germ cells was not measured.
DISCUSSION
In 1979, Sillence et al.1 recognized both dominant and recessive modes of inheritance in families with OI and thought that the perinatal lethal type, OI type II, was recessively inherited, an insight that has proved to be partially correct. Counseling of families at risk for recurrence of lethal genetic disorders in the context of simple Mendelian inheritance is straightforward. For dominant lethal disorders, the risk for unaffected parents to have a second affected child depends on whether one parent is mosaic for the mutation and the proportion of germline progenitor cells that carry the mutation. For recessive disorders, the risk to have an affected child after the birth of a first affected child is 25% with each pregnancy.
Evidence that parental germline mosaicism caused recurrence of lethal OI in sibships with unaffected parents arose in the 1980s and made counseling complex.5,8 For a parent with an affected allele in his or her germline, the risk to pass this altered allele depends on the proportion of germ cells that carry the mutant allele. This risk reflects the proportion of cells with the mutant allele allocated to the primordial germline during embryonic development of the parent. The recurrence risk would be 50% to have an affected child with each pregnancy if all allocated cells have one copy of the mutant allele. Generally, parents with germline mosaicism are themselves phenotypically normal (reminiscent of recessive inheritance) and, thus, unaware of the initial risk to have an affected child.
As an aid to prenatal diagnosis and counseling, we set out to determine the proportion of families in which recurrence resulted from parental mosaicism for dominant mutations or autosomal recessive inheritance, the recurrence risk within each of the two groups, whether the mosaicism was more likely to affect one parent than the other, how often mosaicism was detectable in the parents of affected infants, and whether we could estimate the number of cells allocated to the germline.
We found that the primary recurrence rate for perinatal lethal OI, regardless of cause, was 1.3%, similar to what we had recognized more than a decade ago. Among families with a first affected child in which we identified a mutation in a type I collagen gene and studied parental DNA, we documented that in approximately 16% of the families, one parent was mosaic in somatic cells (and in the germline as demonstrated by birth of an affected child).
We studied 37 families in the Repository in which there were two or more affected pregnancies with perinatal lethal OI born to unaffected parents to identify those with dominant mutations and those with recessive mutations. We expected these families to fall into two groups—one in which a parent was mosaic for a dominant mutation in a type I collagen gene and a second in which both parents were carriers for recessive mutations. In the affected infants or pregnancies from 37 families we studied, 26 had dominant mutations in type I collagen genes, 7 had recessive mutations, and we failed to find mutations in the remaining 4 families. In the 26 families in this group with parental germline mosaicism for dominant mutations in type I collagen genes, we studied parental sets in 16 and identified the mutation-bearing cells in somatic sources in 13 of them; in two (one mother and one father), the parent was an obligate mosaic by family structure, and we did not study them. In one, we could not identify the mutant allele in DNA derived from blood from either parent. We can conclude that in the majority of families, the mutation occurred early in parental embryogenesis before allocation of cells to the germline, rather than after allocation. A measured recurrence rate of 27% indicates that on average approximately half of the germ cell precursors carry the mutation and could be consistent with a very small number of cells set aside for the germline. Studies in mice are consistent with a very small precursor pool.21,22 With one exception among those we studied, in DNA from blood in the mosaic individuals, between 10 and 20% of alleles carried the mutation, but this does not serve to refine the recurrence risk or allow any better estimate of the number of cells allocated to the germline.
The measured recurrence rate after the birth of a child with perinatal lethal OI (1.3–2%) is consistent with the observation that approximately 5% of pregnancies with this form of OI result from the recessive forms20 and an overall rate of parental mosaicism of approximately 16% in the families with dominant mutations. In this context, the expected recurrence rate would be close to 4%, similar to what we have observed. In a population with a significantly higher carrier rate for recessive mutations, the predicted recurrence rate would increase in proportion to that rate. If the carrier rate for the West African LEPRE1 mutant allele is 1/200,23 then the calculated recurrence risk, assuming that the rate of parental mosaicism would remain the same, is approximately 8% in that population in which the majority of recurrences would be from the recessively inherited group.
Our study indicates that mutation identification is an important tool for diagnosis and counseling because it allows separation of families into one of three at risk groups: those with a very low risk (well below 0.1% in the absence of parental somatic mosaicism or recessive inheritance), those with a risk of up to 50% in the presence of parental mosaicism (that could be refined by measure of the frequency of the allele in sperm if the father is mosaic), and those with a 25% risk if both parents are carriers of a recessive mutation. The major limitation of this study is that the number of recurrent families is relatively small, and changes in the proportion of recessive forms of OI in the studied population could change the distribution among the three groups. Nonetheless, mutation identification is a key to providing accurate genetic counseling for both early prenatal and preimplantation diagnosis.
REFERENCES
Sillence DO, Senn A, Danks DM . Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979; 16: 101–116.
Young ID, Thompson EM, Hall CM, Pembrey ME . Osteogenesis imperfecta type IIA: evidence for dominant inheritance. J Med Genet 1987; 24: 386–389.
Ramirez F, Sangiorgi FO, Tsipouras P . Human collagens: biochemical, molecular and genetic features in normal and diseased states. Horiz Biochem Biophys 1986; 8: 341–375.
Barsh GS, Roush CL, Bonadio J, Byers PH, Gelinas RE . Intron-mediated recombination may cause a deletion in an alpha 1 type I collagen chain in a lethal form of osteogenesis imperfecta. Proc Natl Acad Sci USA 1985; 82: 2870–2874.
Cohn DH, Byers PH, Steinmann B, Gelinas RE . Lethal osteogenesis imperfecta resulting from a single nucleotide change in one human pro alpha 1(I) collagen allele. Proc Natl Acad Sci USA 1986; 83: 6045–6047.
Byers PH, Starman BJ, Cohn DH, Horwitz AL . A novel mutation causes a perinatal lethal form of osteogenesis imperfecta. An insertion in one alpha 1(I) collagen allele (COL1A1). J Biol Chem 1988; 263: 7855–7861.
Cohn DH, Starman BJ, Blumberg B, Byers PH . Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a dominant mutation in a human type I collagen gene (COL1A1). Am J Hum Genet 1990; 46: 591–601.
Edwards MJ, Wenstrup RJ, Byers PH, Cohn DH . Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a mutation in the COL1A2 gene of type I collagen. The mosaic parent exhibits phenotypic features of a mild form of the disease. Hum Mutat 1992; 1: 47–54.
Young ID, Harper PS . Recurrence risk in osteogenesis imperfecta congenita. Lancet 1980; 1: 432.
Byers PH, Tsipouras P, Bonadio JF, Starman BJ, Schwartz RC . Perinatal lethal osteogenesis imperfecta (OI type II): a biochemically heterogeneous disorder usually due to new mutations in the genes for type I collagen. Am J Hum Genet 1988; 42: 237–248.
Pepin M, Atkinson M, Starman BJ, Byers PH . Strategies and outcomes of prenatal diagnosis for osteogenesis imperfecta: a review of biochemical and molecular studies completed in 129 pregnancies. Prenat Diagn 1997; 17: 559–570.
Morello R, Bertin TK, Chen Y, et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 2006; 127: 291–304.
Barnes AM, Chang W, Morello R, et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med 2006; 355: 2757–2764.
Van Dijk FS, Nesbitt IM, Nikkels PG, et al. CRTAP mutations in lethal and severe osteogenesis imperfecta: the importance of combining biochemical and molecular genetic analysis. Eur J Hum Genet 2009; 17: 1560–1569.
Baldridge D, Schwarze U, Morello R, et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum Mutat 2008; 29: 1435–1442.
Cabral WA, Chang W, Barnes A, et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 2007; 39: 359–365.
Willaert A, Malfait F, Symoens S, et al. Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J Med Genet 2009; 46: 233–241.
van Dijk FS, Nesbitt IM, Zwikstra EH, et al. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet 2009; 85: 521–527.
Barnes AM, Carter EM, Cabral WA, et al. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N Engl J Med 2010; 362: 521–528.
Marini JC, Forlino A, Cabral WA, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 2007; 28: 209–221.
Pace JM, Atkinson M, Willing MC, et al. Deletions and duplications of Gly-Xaa-Yaa triplet repeats in the triple helical domains of type I collagen chains disrupt helix formation and result in several types of osteogenesis imperfecta. Hum Mutat 2001; 18: 319–326.
Bodian DL, Chan TF, Poon A, et al. Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships. Hum Mol Genet 2009; 18: 463–471.
Ohinata Y, Payer B, O'Carroll D, et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature 2005; 436: 207–213.
McLaren A, Lawson KA . How is the mouse germ-cell lineage established?. Differentiation 2005; 73: 435–437.
Marini JC, Cabral WA, Barnes AM . Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res 2010; 339: 59–70.
Acknowledgements
This work was supported in part by Grants from the Osteogenesis Imperfecta Foundation, Inc., a student fellowship from Grad into Med Molecular Medicine program of the Howard Hughes Medical Institute at the University of Washington, National Institutes of Health (AR051582), and the Freudmann Fund for Translational Research in Ehlers Danlos syndrome at the University of Washington. The authors thank the clinicians and families who contributed samples and clinical details to allow this analysis to be done.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Pyott, S., Pepin, M., Schwarze, U. et al. Recurrence of perinatal lethal osteogenesis imperfecta in sibships: Parsing the risk between parental mosaicism for dominant mutations and autosomal recessive inheritance. Genet Med 13, 125–130 (2011). https://doi.org/10.1097/GIM.0b013e318202e0f6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e318202e0f6
Keywords
This article is cited by
-
Co-occurrence of heterozygous mutations in COL1A1 and SERPINF1 in a high-risk pregnancy complicated by osteogenesis imperfecta
Journal of Genetics (2019)
-
Parental mosaicism is a pitfall in preimplantation genetic diagnosis of dominant disorders
European Journal of Human Genetics (2014)
-
EMQN best practice guidelines for the laboratory diagnosis of osteogenesis imperfecta
European Journal of Human Genetics (2012)
-
Osteogenesis imperfecta
Medizinische Genetik (2012)
