
Abstract
Purpose:
Thoracic aortic aneurysm and dissection (TAAD) is typically inherited in an autosomal dominant manner, but rare X-linked families have been described. So far, the only known X-linked gene is FLNA, which is associated with the periventricular nodular heterotopia type of Ehlers-Danlos syndrome. However, mutations in this gene explain only a small number of X-linked TAAD families.
Methods:
We performed targeted resequencing of 368 candidate genes in a cohort of 11 molecularly unexplained Marfan probands. Subsequently, Sanger sequencing of BGN in 360 male and 155 female molecularly unexplained TAAD probands was performed.
Results:
We found five individuals with loss-of-function mutations in BGN encoding the small leucine-rich proteoglycan biglycan. The clinical phenotype is characterized by early-onset aortic aneurysm and dissection. Other recurrent findings include hypertelorism, pectus deformity, joint hypermobility, contractures, and mild skeletal dysplasia. Fluorescent staining revealed an increase in TGF-β signaling, evidenced by an increase in nuclear pSMAD2 in the aortic wall. Our results are in line with those of prior reports demonstrating that Bgn-deficient male BALB/cA mice die from aortic rupture.
Conclusion:
In conclusion, BGN gene defects in humans cause an X-linked syndromic form of severe TAAD that is associated with preservation of elastic fibers and increased TGF-β signaling.
Genet Med 19 4, 386–395.
Similar content being viewed by others

Introduction
Thoracic aortic aneurysms (TAA) are often asymptomatic but predispose to aortic dissections, which are associated with high mortality rates.1 TAAs and dissections (TAAD) can be subdivided into syndromic (associated with systemic manifestations) and nonsyndromic forms. Most commonly, familial TAADs segregate in an autosomal dominant manner, but rare X-linked families have been described.2 So far, FLNA is the only X-linked gene associated with a syndromic form of TAAD, namely, periventricular nodular heterotopia type 1 (PVNH1; also known as the Ehlers-Danlos variant of PVNH, MIM300049). However, FLNA explains only a small number of the X-linked TAAD families.3
Marfan syndrome (MFS; MIM154700) is an autosomal dominant connective tissue disorder; affected individuals present with ocular, skeletal, and cutaneous signs in addition to aneurysm and dissection. Loeys-Dietz syndrome (LDS; MIM609192, MIM610168, MIM613795, MIM614816, MIM615582, and MIM601366) is an aneurysmal connective tissue disorder that can be distinguished from MFS by the unique presence of craniofacial, skeletal, cutaneous, and/or vascular manifestations and prominently includes hypertelorism, cleft palate, or bifid uvula and arterial tortuosity, with aneurysms distant from the aortic root. In addition, aneurysms in individuals with LDS tend to dissect at an earlier age and smaller diameter compared to individuals with MFS.4 Whereas MFS is caused by mutations in FBN1 coding for an extracellular matrix (ECM) protein,5 LDS is caused by loss-of-function (LOF) mutations in genes coding for components of the transforming growth factor β (TGF-β) signaling pathway (TGFBR1/2, SMAD2/3, TGFB2/3).6 Recent work has demonstrated that both MFS and LDS lead to dysregulation of the TGF-β signaling pathway.4,7
Biglycan deficiency in male BALB/cA mice has been shown to lead to sudden death due to aortic rupture, suggesting that biglycan might be essential for the structural integrity of the aortic wall. Additionally, BGN mRNA and protein expression are reduced in individuals with Turner syndrome (45,X),8 who suffer more frequently from vascular anomalies including aortic dissection and rupture.9 Human mutations in BGN have not been described in TAAD probands.
Here, we report that BGN mutations in humans cause an X-linked, severe, syndromic form of TAAD, including hypertelorism, pectus deformity, joint hypermobility, contractures, and mild skeletal dysplasia.
Materials and Methods
Human participants, DNA, and aortic specimens
The study was approved by the appropriate institutional ethics review boards, and the required informed consent was obtained from all participating subjects. Additionally, informed consent for the publication of photos was obtained from the subjects or legal guardians. DNA of additional affected and unaffected family members was requested whenever it was considered informative. Aortic specimens from probands 3-III-2 and 5-II-1 as well as from two gender-matched control samples obtained from deceased individuals who did not have cardiovascular diseases were at our disposal. As positive controls, we used available aortic specimens from two individuals with LDS (TGFB3 mutation, p.Asp263His). Fibroblasts were cultured using skin biopsy specimens from probands 4-II-1 and 5-II-1 and two gender-matched control individuals.
Targeted resequencing
We performed targeted resequencing of 368 ECM-related and TGF-β-related genes using the HaloPlex Target Enrichment System (Agilent Technologies, Santa Clara, CA). Samples were paired-end sequenced using 2 × 100 bp on a HiSeq1500 in high-output mode (Illumina, San Diego, CA). Enrichment, data analysis, variant annotation, and confirmation of the variants were performed as previously described.10,11 GRCh37 was used as a reference build.
Sanger sequencing and microarray
Sanger sequencing of the seven coding exons of BGN (RefSeq transcript NM_001711.3) was performed. The PCR primer sequences and reaction conditions are listed in Supplementary Table S1 online. PCR products were bidirectionally sequenced using the BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Carlsbad, CA) and separated on an ABI 3130XL Genetic Analyzer (Applied Biosystems). Sequences were analyzed using CLC DNA workbench (CLC Bio, Aarhus, Denmark). Microarray analysis was performed using the Illumina HumanCytoSNP12-V2.1 BeadChip (Illumina, San Diego, CA) according to standard protocols. Copy-number variants (CNVs) were analyzed using CNV-WebStore.12
Cell culture
Skin fibroblasts were cultured in Roswell Park Memorial Institute medium and supplemented with 15% fetal bovine serum, 1% L-glutamine, 1% sodium pyruvate, 1% Penicillin/Streptomycin, and 0.1% Primocin. Experiments were performed at passages 2 to 4. The fibroblasts were incubated with and without puromycin (200 µg/ml) to inhibit nonsense-mediated decay (NMD).
cDNA sequencing and cloning
RNA was extracted from skin fibroblasts (from proband 5-II-1) to identify changes in BGN splicing using the RNeasy mini kit (Qiagen, Valencia, CA), followed by random hexamer cDNA conversion with the Superscript III First-Strand Synthesis kit for RT-PCR (Invitrogen, ThermoFisher Scientific, Waltham, MA). PCR was performed on the obtained cDNA using the following primers: 5′-CAGAGAGGCTTCTGGGACTT-3′ and 5′-GACGAGGGCGTAGAGGTG-3′. The resulting PCR product was then cloned into One Shot TOP10 cells (ThermoFisher Scientific), after which 94 colonies were picked. The different splice products were sequenced with Sanger sequencing.
Histology and immunohistochemistry
Paraffin-embedded aortic tissue of affected individuals and of control samples were sectioned (5 µm) with the HM340E Microm microtome (ThermoFisher Scientific). Histological staining, including Verhoeff-Van Gieson and Masson’s Trichrome, was performed using standard protocols.
Fluorescent staining of the aortic tissue with primary antibodies against biglycan (AF2667, 1:40; R&D Systems, Minneapolis, MN), decorin (AF143, 1:40: R&D Systems), and pSMAD2 (04-953, 1:100; Millipore, Billerica, MA) was performed as previously described13 but with the following modifications: the secondary antibodies included chicken-anti-goat Alexa Fluor 488 conjugate (A-21467, 1:200; Life Technologies, ThermoFisher Scientific) and goat-anti-rabbit TRITC conjugate (T-2769, 1:200; Life Technologies, ThermoFisher Scientific), and treatment with the Fc block reagent was omitted from the protocol. For immunohistochemistry with anti-biglycan and anti-decorin, a pretreatment step with chondroitin ABC lyase (1.25 U/ml, C3667; Sigma-Aldrich, St. Louis, MO) was performed after deparaffinization.14 Confocal images were acquired using the UltraView imaging analysis system (PerkinElmer, Waltham, MA) at 20× and 40× magnification. The number of pSMAD2-positive nuclei was counted in eight fields (40× magnification) for the media and in four fields (40× magnification) for the adventitia by five blinded observers.
Results
Identification of BGN mutations in syndromic TAAD probands
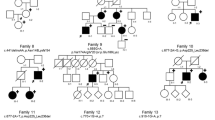
After targeted resequencing of 368 ECM-related and TGF-β-related genes in a cohort of 11 molecularly unexplained Marfan probands, we identified two individuals with mutations in BGN encoding the ECM small leucine-rich proteoglycan (SLRP) biglycan. In the proband (1-III-1) of family 1 ( Figure 1a ), we identified a novel nonsense mutation at amino acid position 2 (c.5G>A, p.Trp2*, ClinVar accession number SCV000266568), and we identified a novel missense variant at the exon–intron boundary of exon 7 (A in AGgt of the donor splice site) in the proband (2-II-1) of family 2 (c.908A>C, p.Gln303Pro, ClinVar accession number SCV000266569). Only a minor effect was predicted on splicing (Supplementary Table S2 online), but no skin fibroblasts of the proband of family 2 were available to investigate potential aberrant splicing. However, the mutation substitutes a highly conserved amino acid ( Figure 1c , Supplementary Table S2 online).

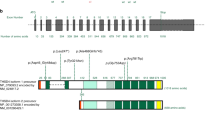
Mutation analysis and BGN structure. (a) Pedigrees of the families with their respective mutations. Squares represent males, circles represent females, filled symbols represent aortic aneurysm/dissection and/or systemic involvement, half-filled symbols represent individuals with incomplete clinical data, and + or − sign denotes presence or absence of BGN mutation. The brackets represent an individual who was adopted into the family. (b) Location of BGN on the X chromosome and identified mutations. Deletions are marked with a red bar. ATP2B3 isoform 1 represents ENST00000359149. ATP2B2 isoforms 2 and 3 represent ENST00000370186 and ENST00000349466. (c) Conservation of residues on amino acid position 80 and position 303 in other species and in human DCN.
Subsequently, we performed Sanger sequencing of BGN in 360 male and 155 female TAAD probands negative for mutations in the known TAAD genes.10 Because we were unable to amplify exons 2 to 8 of the BGN gene in the male probands (3-III-1 en 4-II-1) of families 3 and 4 ( Figure 1a ), we suspected a partial hemizygous gene deletion. The latter was confirmed by microarray analysis ( Figure 1 ).We further delineated these deletions by PCR and Sanger sequencing across the breakpoints. In family 3, we identified a 21-kb deletion spanning chrX:152767424-152787984 (ClinVar accession number SCV000266570) and comprising exons 2 to 8 of BGN. In family 4, we identified a larger deletion comprising 28 kb and spanning chrX:152768438-152795976 (ClinVar accession number SCV000266571), but also deleting exons 2 to 8 of BGN. In the male proband (5-II-1) of family 5 ( Figure 1a ), we identified a novel missense mutation at the exon–intron boundary of exon 2, which was predicted to lead to aberrant splicing because the donor splice site was affected (c.238G>A, p.Gly80Ser, ClinVar accession number SCV000266572, Supplementary Table S2 online).
We subsequently sequenced cDNA from skin fibroblasts of proband 5-II-1 cultured with and without puromycin, a translational inhibitor that will stabilize out-of-frame transcripts that are normally destined to undergo nonsense-mediated mRNA decay (NMD). Three alternatively spliced products were observed ( Figure 2 ). Of the total amount of BGN transcript, only 8% shows normal splicing. The first alternative product (4% representation) incorporates 45 bp of the intron without creating a premature stop codon. The remaining two alternatively spliced products incorporate 111 and 368 bp of the intron (with 68 and 20% representation, respectively) and do introduce a premature stop codon leading to NMD.

Aberrant splicing in family 5. In the proband of family 5, three aberrantly spliced mRNAs are produced. The cDNA (converted after inhibition of nonsense-mediated decay with puromycin) was cloned into a vector and the number of colonies with the different products was counted (total counted n = 94).
In conclusion, all identified mutations are predicted to lead to a partial or complete loss of function of BGN through absence of a functional copy of the gene, through NMD, or through predicted functional changes to protein function (Supplementary Table S3 online).
Clinical data
The clinical phenotype of the individuals ( Figure 3 ) with BGN mutations is characterized by early-onset aortic aneurysm (as early as age 1; 4-II-1) and dissection (earliest at the age of 15; 3-III-1) in male probands. In all five families, either the aortic root or the more distal ascending aorta was involved. In one family, we also observed brain aneurysms in one individual (5-I-2; Figure 1a ). Although the mitral and aortic valves were involved in some families (e.g., 2-III-1 and 4-I-2), insufficiency was mild. The cardiovascular phenotype in mutation-carrying females ranged from unaffected upon repeated echocardiographic evaluation (3-II-2 and 3-I-2) over aortic root dilatation (4-I-2 and 5-I-2) to death due to aortic dissection (1-II-1). The initial suspected clinical (differential) diagnoses varied between MFS (families 1 and 2), LDS (families 3, 4, 5), and Melnick-Needles syndrome and filaminopathy (families 3 and 4).

Clinical features. (a) Clinical features include hypertelorism, malar flattening, down-slanting palpebral fissures (3-II-2/3/4, 3-III-1/2, and 4-II-1), proptosis (3-III-1/2), joint hypermobility (4-II-1), short spatulous fingers (3-III-1/2), and camptodactyly of fingers and toes (3-III-1/2). (b) Skeletal survey of patient 3-III-1 at age 10 years and of patient 4-II-1 at age 1.5 years. Radiograph of both hands (3-III-1) reveals asymmetry in the size of the carpal bones, with the ones on the right being larger. Overall, there is symmetric shortening and broadening of the metacarpals and phalanges that appear osteopenic with relatively thin cortices. Lateral view of the spine (3-III-1) demonstrates platyspondyly with thoracolumbar transition mild anterior tonguing. Anteroposterior view of the lower limbs (3-III-1) shows that the distal (sub)metaphyseal region of the femur is widened and the knee epiphyses are flattened. There is also mild undermodeling of the tibia with an S-shape configuration of the diaphysis. The distal tibial epiphysis is dysplastic. Skeletal radiographs of proband 4-II-1 reveal bilateral hip dislocation (treated conservatively), broadened and triangular middle phalanges, broad terminal phalanges, and a broad thorax.
We observed nonspecific connective tissue features (e.g., pectus deformities, joint hypermobility/contractures, and striae) and more specific manifestations of LDS (e.g., bifid uvula, hypertelorism, and cervical spine instability) in affected individuals. The stature of the affected patients in this series ranged from short (e.g., 3-III-2 and 4-II-1) to normal to high normal (e.g., 1-III-1, 2-III-1 and 5-II-1) when compared to unaffected family members. Similarly, either long fingers (families 1 and 2) or rather short, broad fingers (families 3 and 4, 5-I-2; Figure 3 ) were noted. Other unusual features not typically seen in MFS or LDS include ventricular dilatation on brain imaging (families 3 and 4), hypertrichosis (family 3), gingival hypertrophy (family 4), and relative macrocephaly (families 3 and 4). There is also evidence of skeletal dysplasia with hip dislocation (families 3 and 4), platyspondyly (family 3), phalangeal dysplasia (families 3 and 4), and dysplastic epiphyses of the long bones (family 3; Figure 3 ). A detailed clinical description of the five families can be found in the Supplementary Data online.
Structural and functional changes in the aortic wall
To investigate the collagen and elastic fiber contents of the aortic walls, we stained aortic walls from affected (3-III-2 and 5-II-1) and control individuals with Verhoeff-Van Gieson and Trichrome Masson. The staining revealed low to normal collagen content; elastin fibers appeared normal ( Figure 4a – h ). This is in contrast to what is observed in LDS individuals, in whom an increase in collagen production and fragmentation of elastic fibers are typically present.13,15

Histological and fluorescent staining. (a–d) Verhoeff-Van Gieson staining of the control sample (a), proband 3-III-2 (b), proband 5-II-1 (c), and an individual with LDS (d). The elastin fibers in proband 3-III-2 do not show any clear breaks. In the tissue from the individual with LDS, a clear reduction in the elastic fiber content is present. Scale bar indicates 5 mm. (e–h) Trichrome Masson staining of the control sample (e), proband 3-III-2 (f), proband 5-II-1 (g), and an individual with LDS (h). A clear reduction of collagen content is present in proband 3-III-2, whereas an increase is observed in the individual with LDS. (i–l) Biglycan staining of the control sample (i), proband 3-III-2 (j), proband 5-II-1 (k), and an individual with LDS (l). Biglycan is not expressed in proband 3-III-2, whereas in proband 5-II-1 some residual expression of biglycan is present. In the proband with LDS, focal increased expression of biglycan can be observed. Scale bar indicates 25 µm. (m–p) pSMAD2 staining of the control sample (m), proband 3-III-2 (n), proband 5-II-1 (o), and an individual with LDS (p). No pSMAD2-positive nuclei are present in the control sample. In proband 3-III-2, proband 5-II-1, and the proband with LDS, an increase in pSMAD2-positive nuclei can be observed.
Next, we fluorescently stained the aortic tissue of the affected probands for biglycan ( Figure 4i – l ). As expected, no protein expression was detected in proband 3-III-2 because this proband had a deletion of BGN. We observed a subtle expression of biglycan in proband 5-II-1 that was related to the predicted expression of a small amount of wild-type (WT) spliced protein. Interestingly, in individuals with LDS (TGFB3 mutation, p.Asp263His), we specifically observed expression of biglycan in areas with disorganization of the arterial structure. The latter is different than what has been described before in patients with ascending aortic aneurysms of unknown genetic origin.16
To investigate whether decorin expression was altered in BGN-deficient patients, we performed immunohistological staining of the aortic walls. Decorin is another member of the class I SLRP, which displays an essential role in the formation and deposition of collagen fibrils and was previously suggested to be able to compensate for biglycan loss.17 We confirmed focal expression of decorin in the media of the aortic walls of individuals with a BGN mutation (Supplementary Figure S1 online) that was comparable to what has been described in aortic aneurysm patients.16
Because BGN has also been suggested to be a regulator of TGF-β signaling,18,19 and because we observed marked phenotypic similarities with MFS and LDS, we hypothesized that an increase in TGF-β signaling could also underlie the phenotype seen in individuals with a BGN mutation. Therefore, we stained pSMAD2, a specific marker of activated canonical TGF-β signaling ( Figure 4m – p ). We observed an increase of pSMAD2-positive nuclei compared to controls, thus confirming an increase in TGF-β signaling. In addition, a gradient of increased nuclear pSMAD2 staining toward the adventitia was observed in proband 3-III-2 (Supplementary Figure S2 online). The increased TGF-β signaling in the media was comparable to the LDS-affected individual.
Discussion
The BGN gene is located on chromosome Xq28 and encodes the biglycan protein.20 Biglycan belongs to the SLRP class I proteins and, together with other proteoglycans such as decorin, is mainly involved in ECM assembly and maintenance.21 It consists of a small protein core (~42 kDa)14 that contains 10 leucine-rich repeats (LRR)22 with two tissue-specific chondroitin or dermatan-sulfate glycosaminoglycan (GAG) chains attached to two Ser-Gly sites in the N-terminus of the core protein.23,24 Through the core protein and GAG chains, biglycan interacts with many other ECM proteins, including collagen type I, II, III, and VI and elastin.25,26,27,28 In this manner, biglycan becomes sequestered in the ECM of most organs; its role seems to be not only that of a mechanical link between matrix components but also that of growth factor binding and regulation of signaling. In healthy individuals, biglycan is expressed by various specialized cell types, including endothelial cells, skeletal myocytes, and differentiating keratinocytes.14 It is widely expressed in various tissues throughout the body, including bone, skin, heart, lung, and arteries.14,29 In the aorta, biglycan is present in the intima and the media,14 but the adventitia is the major site of deposition.17
Due to the suggested structural role of biglycan, we performed histological staining to assess the architecture of the aortic wall. We observed a normal appearance of elastic fibers and low to normal collagen content in the aortic wall of our probands. This is in contrast to what has been described for MFS and LDS aortic tissue, where an increase in collagen content and an increase in elastic fiber breaks are typically observed.15,30,31 As observed in bgn-deficient mice, we hypothesize that proper functioning of biglycan is necessary for correct collagen fibril diameters and lateral association of fibers rather than collagen amount.32
In addition to its structural role, biglycan interacts with several growth factors and cytokines, including TGF-β,18,19 to modulate proliferation, migration, and differentiation of cells.33 This regulatory function resembles that of fibrillin-1, which also forms a reservoir for latent TGF-β.7 Our results indicate that lack of biglycan increases TGF-β signaling, especially in the adventitia, the major site of biglycan deposition in the aortic wall.17 This may explain why BGN deficiency has a more pronounced effect on TGF-β signaling in this layer of the aortic wall.17 Until now, the upregulation of TGF-β signaling in other syndromic forms of TAAD, like MFS and LDS, has mainly been studied in the media of the aortic wall.15 In individuals with a BGN mutation, we observed a slight increase in TGF-β signaling in the media; however, this increase was significantly more pronounced in the adventitia (Supplementary Figure S2 online). The increase in TGF-β signaling is expected to lead to an increased transcription of collagen, but proper collagen fibril assembly may be hampered by biglycan deficiency. Our findings of increased TGF-β signaling are in line with prior observations of enhanced SMAD2 phosphorylation in bgn+/− cardiac fibroblasts.34 The latter induced differentiation into a pro-proliferative myofibroblast phenotype that is rescued by TGF-β neutralizing antibodies.
The absence of an aortic phenotype in the pure C57Bl6 Bgn-deficient mice35 confirms our hypothesis of increased TGF-β signaling as the driving pathogenetic mechanism of the aortic aneurysm development. The C57Bl6 mouse background is believed to induce a relative deficiency state for TGF-β signaling and, as such, attenuates high TGF-β signaling in Bgn-deficient aortic walls. Only in male BALB/cA mice does biglycan deficiency lead to sudden death from aortic rupture, indicating that biglycan is both structurally and functionally essential for the integrity of the aortic wall.17 The molecular mechanism for the difference between the pure C57Bl6 and the BALB/cA backgrounds remains unknown.
As a consequence of the mice studies discussed, biglycan gene defects were predicted to play a potential role in the pathogenesis of aortic dissection and rupture in humans.17 Yet, this study is the first to report BGN mutations in TAAD probands. In Bgn knockout mice, aortic elastic lamella did not display any structural changes,17 similar to the human BGN probands. These normal elastic fibers can potentially be explained by the ability of decorin to substitute for biglycan in pathological contexts.17 Decorin is another member of the class I SLRP, which displays an essential role in the formation and deposition of collagen fibrils and, like biglycan, is able to bind tropoelastin, fibrillin-1, and microfibril-associated glycoprotein 1.26,36 However, its expression pattern is somewhat different from that of biglycan and, depending on the tissue, its synergistic or additive effects with biglycan can differ.37 Bgn/Dcn double-knockout mice on a 129Sv/C57BL6 background have been generated, but an aortic phenotype has not been extensively studied.37 As such, it might be interesting to study the aortic phenotype of the Bgn/Dcn double-knockout mice on a BALB/cA background.
In addition to their cardiovascular phenotype, Bgn-deficient mice have also been reported to show a phenotype characterized by growth failure, reduced bone formation, and age-related severe osteopenia.32 Previous research has indicated that the expression levels of biglycan are potentially related to stature in humans: patients with Turner syndrome have low levels of biglycan and typically display short stature, whereas patients with triple X syndrome present with increased limb length and high levels of biglycan.32 In families 3 and 4, affected male individuals presented short stature ( Table 1 ) and mild skeletal dysplasia ( Figure 3b ), confirming a role for biglycan in human bone development.
From a clinical perspective, the phenotype of BGN-mutated individuals overlaps with LDS and MFS individuals. Typical LDS features in families 3 and 4 include pronounced hypertelorism, bifid uvula, and early-onset aortic dissections. The probands of families 1 and 2 were initially diagnosed with MFS, but they also displayed LDS-like features such as hypertelorism. The phenotype of the carrier females ranges from unaffected upon repeated echocardiographic examination (3-II-2 and 3-I-2) over aortic root dilatation (4-I-2 and 5-I-2) to death due to aortic dissection (1-II-1). Overall, the noncardiovascular characteristics of the female carriers seem milder. To determine whether skewed X inactivation underlies the difference between affected and unaffected females, we performed X-inactivation experiments using the HUMARA assay.38 The results did not reveal a clear pattern of skewed X inactivation that could explain the observed difference (data not shown).
The phenotypic features in families 3 and 4 seemed more pronounced than in the other families. Complete LOF cannot explain this difference between the families because we also expect a complete LOF in family 1 due to the stop codon at the second amino acid position (even if the first downstream ATG would be used as an alternative start codon, the resulting protein would miss the signal and the propeptide of biglycan, and thus still lead to LOF). However, the deletion occurring in families 3 and family 4 extends beyond exons 2–8 of BGN and includes a region downstream of the BGN gene that contains a CpG island and, in addition, the untranslated region (5ʹ-UTR) of two alternative protein coding transcripts of the downstream gene ATP2B3. So far, mutations in ATP2B3 are linked to an X-linked form of spinocerebellar ataxia39,40 and therefore cannot be directly linked to the phenotype of the BGN probands. Nonetheless, (intronic) regulatory elements may play a role in the expression of the phenotype and explain the observed difference. The milder phenotype in family 5 could be correlated with 8% residual expression of WT spliced biglycan protein.
In conclusion, our results confirm that BGN gene defects in humans cause an X-linked syndromic form of severe TAAD. The identification of mutations in BGN contributes to the molecular diagnosis of X-linked TAAD and should therefore be implemented in diagnostics. The signature of increased TGF-β signaling in the aortic walls of the BGN-deficient patients offers an interesting therapeutic target for TGF-β activity attenuating agents such as angiotensin receptor blockers.
Disclosure
The authors declare no conflict of interest.
References
Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011;473:308–316.
Coady MA, Davies RR, Roberts M, et al. Familial patterns of thoracic aortic aneurysms. Arch Surg 1999;134:361–367.
Sheen VL, Jansen A, Chen MH, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 2005;64:254–262.
Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005;37:275–281.
Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991;352:337–339.
Verstraeten A, Alaerts M, Van Laer L, Loeys B. Marfan syndrome and related disorders: 25 years of gene discovery. Hum Mutat 2016;37 :524–531.
Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet 2003;33:407–411.
Geerkens C, Vetter U, Just W, et al. The X-chromosomal human biglycan gene BGN is subject to X inactivation but is transcribed like an X-Y homologous gene. Hum Genet 1995;96:44–52.
Lin AE, Lippe BM, Geffner ME, et al. Aortic dilation, dissection, and rupture in patients with Turner syndrome. J Pediatr 1986;109:820–826.
Proost D, Vandeweyer G, Meester JA, et al. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum Mutat 2015;36:808–814.
Vandeweyer G, Van Laer L, Loeys B, Van den Bulcke T, Kooy RF. VariantDB: a flexible annotation and filtering portal for next generation sequencing data. Genome Med 2014;6:74.
Vandeweyer G, Reyniers E, Wuyts W, Rooms L, Kooy RF. CNV-WebStore: online CNV analysis, storage and interpretation. BMC Bioinformatics 2011;12:4.
Bertoli-Avella AM, Gillis E, Morisaki H, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol 2015;65:1324–1336.
Bianco P, Fisher LW, Young MF, Termine JD, Robey PG. Expression and localization of the two small proteoglycans biglycan and decorin in developing human skeletal and non-skeletal tissues. J Histochem Cytochem 1990;38:1549–1563.
Maleszewski JJ, Miller DV, Lu J, Dietz HC, Halushka MK. Histopathologic findings in ascending aortas from individuals with Loeys-Dietz syndrome (LDS). Am J Surg Pathol 2009;33:194–201.
Gomez D, Al Haj Zen A, Borges LF, et al. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J Pathol 2009;218:131–142.
Heegaard AM, Corsi A, Danielsen CC, et al. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation 2007;115:2731–2738.
Hildebrand A, Romarís M, Rasmussen LM, et al. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J 1994;302 (Pt 2):527–534.
Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol 2001;280:L1327–L1334.
McBride OW, Fisher LW, Young MF. Localization of PGI (biglycan, BGN) and PGII (decorin, DCN, PG-40) genes on human chromosomes Xq13-qter and 12q, respectively. Genomics 1990;6:219–225.
Halper J. Proteoglycans and diseases of soft tissues. Adv Exp Med Biol 2014;802:49–58.
Iozzo RV. The family of the small leucine-rich proteoglycans: key regulators of matrix assembly and cellular growth. Crit Rev Biochem Mol Biol 1997;32:141–174.
Choi HU, Johnson TL, Pal S, Tang LH, Rosenberg L, Neame PJ. Characterization of the dermatan sulfate proteoglycans, DS-PGI and DS-PGII, from bovine articular cartilage and skin isolated by octyl-sepharose chromatography. J Biol Chem 1989;264:2876–2884.
Roughley PJ, White RJ. Dermatan sulphate proteoglycans of human articular cartilage. The properties of dermatan sulphate proteoglycans I and II. Biochem J 1989;262:823–827.
Douglas T, Heinemann S, Bierbaum S, Scharnweber D, Worch H. Fibrillogenesis of collagen types I, II, and III with small leucine-rich proteoglycans decorin and biglycan. Biomacromolecules 2006;7:2388–2393.
Reinboth B, Hanssen E, Cleary EG, Gibson MA. Molecular interactions of biglycan and decorin with elastic fiber components: biglycan forms a ternary complex with tropoelastin and microfibril-associated glycoprotein 1. J Biol Chem 2002;277:3950–3957.
Schönherr E, Witsch-Prehm P, Harrach B, Robenek H, Rauterberg J, Kresse H. Interaction of biglycan with type I collagen. J Biol Chem 1995;270:2776–2783.
Wiberg C, Heinegård D, Wenglén C, Timpl R, Mörgelin M. Biglycan organizes collagen VI into hexagonal-like networks resembling tissue structures. J Biol Chem 2002;277:49120–49126.
Yeo TK, Torok MA, Kraus HL, Evans SA, Zhou Y, Marcum JA. Distribution of biglycan and its propeptide form in rat and bovine aortic tissue. J Vasc Res 1995;32:175–182.
Trotter SE, Olsen EG. Marfan’s disease and Erdheim’s cystic medionecrosis. A study of their pathology. Eur Heart J 1991;12:83–87.
Collins MJ, Dev V, Strauss BH, Fedak PW, Butany J. Variation in the histopathological features of patients with ascending aortic aneurysms: a study of 111 surgically excised cases. J Clin Pathol 2008;61:519–523.
Xu T, Bianco P, Fisher LW, et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat Genet 1998;20:78–82.
Lawrence DA. Transforming growth factor-beta: a general review. Eur Cytokine Netw 1996;7:363–374.
Melchior-Becker A, Dai G, Ding Z, et al. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J Biol Chem 2011;286:17365–17375.
Tang T, Thompson JC, Wilson PG, Nelson C, Williams KJ, Tannock LR. Decreased body fat, elevated plasma transforming growth factor-β levels, and impaired BMP4-like signaling in biglycan-deficient mice. Connect Tissue Res 2013;54:5–13.
Trask BC, Trask TM, Broekelmann T, Mecham RP. The microfibrillar proteins MAGP-1 and fibrillin-1 form a ternary complex with the chondroitin sulfate proteoglycan decorin. Mol Biol Cell 2000;11:1499–1507.
Corsi A, Xu T, Chen XD, et al. Phenotypic effects of biglycan deficiency are linked to collagen fibril abnormalities, are synergized by decorin deficiency, and mimic Ehlers-Danlos-like changes in bone and other connective tissues. J Bone Miner Res 2002;17:1180–1189.
Boudewijns M, van Dongen JJ, Langerak AW. The human androgen receptor X-chromosome inactivation assay for clonality diagnostics of natural killer cell proliferations. J Mol Diagn 2007;9:337–344.
Bertini E, des Portes V, Zanni G, et al. X-linked congenital ataxia: a clinical and genetic study. Am J Med Genet 2000;92:53–56.
Zanni G, Calì T, Kalscheuer VM, et al. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc Natl Acad Sci USA 2012;109:14514–14519.
Acknowledgements
We are grateful to the families who participated in this study. We acknowledge Christophe Hermans (CORE, University of Antwerp) and Dominique De Rijck (Department of Cell Biology and Histology, University of Antwerp) for technical support. This research was supported by funding from the University of Antwerp (Lanceringsproject), the Fund for Scientific Research, Flanders (FWO, Belgium, G.0221.12), the Dutch Heart Foundation (2013T093), the Fondation Leducq (MIBAVA–Leducq 12CVD03), and an ancillary genetics grant to the NIH Marfan losartan study from the National Marfan Foundation. B.L.L. is senior clinical investigator of the Fund for Scientific Research, Flanders, and holds a starting grant from the European Research Council (ERC- StG-2012-30972-BRAVE). J.A.N.M. is a predoctoral researcher of the Fund for Scientific Research, Flanders (FWO, Belgium).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
(DOCX 1215 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Meester, J., Vandeweyer, G., Pintelon, I. et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet Med 19, 386–395 (2017). https://doi.org/10.1038/gim.2016.126
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2016.126
Keywords
This article is cited by
-
Expanding the clinical spectrum of biglycan-related Meester-Loeys syndrome
npj Genomic Medicine (2024)
-
Sex-dependent differences in the secretome of human endothelial cells
Biology of Sex Differences (2021)
-
Sequence features, structure, ligand interaction, and diseases in small leucine rich repeat proteoglycans
Journal of Cell Communication and Signaling (2021)
-
Biglycan: an emerging small leucine-rich proteoglycan (SLRP) marker and its clinicopathological significance
Molecular and Cellular Biochemistry (2021)
-
Transforming Growth Factor-β and the Renin-Angiotensin System in Syndromic Thoracic Aortic Aneurysms: Implications for Treatment
Cardiovascular Drugs and Therapy (2021)
