
Abstract
Classic Ehlers-Danlos syndrome is a heritable connective tissue disorder characterized by skin hyperextensibility, fragile and soft skin, delayed wound healing with formation of atrophic scars, easy bruising, and generalized joint hypermobility. It comprises Ehlers-Danlos syndrome type I and Ehlers-Danlos syndrome type II, but it is now apparent that these form a continuum of clinical findings and differ only in phenotypic severity. It is currently estimated that approximately 50% of patients with a clinical diagnosis of classic Ehlers-Danlos syndrome harbor mutations in the COL5A1 and the COL5A2 gene, encoding the α1 and the α2-chain of type V collagen, respectively. However, because no prospective molecular studies of COL5A1 and COL5A2 have been performed in a clinically well-defined patient group, this number may underestimate the real proportion of patients with classic Ehlers-Danlos syndrome harboring a mutation in one of these genes. In the majority of patients with molecularly characterized classic Ehlers-Danlos syndrome, the disease is caused by a mutation leading to a nonfunctional COL5A1 allele and resulting in haploinsufficiency of type V collagen. A smaller proportion of patients harbor a structural mutation in COL5A1 or COL5A2, causing the production of a functionally defective type V collagen protein. Most mutations identified so far result in a reduced amount of type V collagen in the connective tissues available for collagen fibrillogenesis. Inter- and intrafamilial phenotypic variability is observed, but no genotype-phenotype correlations have been observed. No treatment for the underlying defect is presently available for Ehlers-Danlos syndrome. However, a series of preventive guidelines are applicable.
Similar content being viewed by others

CLINICAL DESCRIPTION
Prevalence and clinical diagnosis
Classic Ehlers-Danlos syndrome (EDS) is a heritable connective tissue disorder characterized mainly by skin hyperextensibility, abnormal wound healing, and joint hypermobility. The prevalence of classic EDS has been estimated to be 1:20,000.1 It includes two previously designated subtypes (EDS type I or “gravis type” and EDS type II or “mitis type”) that are now recognized to form a clinical continuum. It is likely that some individuals with milder manifestations of the disease, previously classified as EDS type II, do not come to medical attention and, therefore, go undetected.
The diagnosis of EDS, classic type is established by clinical examination and family history. Diagnostic criteria were developed by a medical advisory group in a conference at Villefranche in 1997.2 The combination of the three major diagnostic criteria is highly specific for the presence of the condition:
Table 1 Beighton criteria for joint hypermobility
-
Skin hyperextensibility: Skin hyperextensibility (Fig. 1) should be tested at a neutral site (one not subjected to mechanical forces or scarring), such as the volar surface of the forearm. It is measured by pulling up the skin until resistance is felt. In young children, hyperextensibility of the skin is difficult to assess because of abundant subcutaneous fat.
Fig. 1 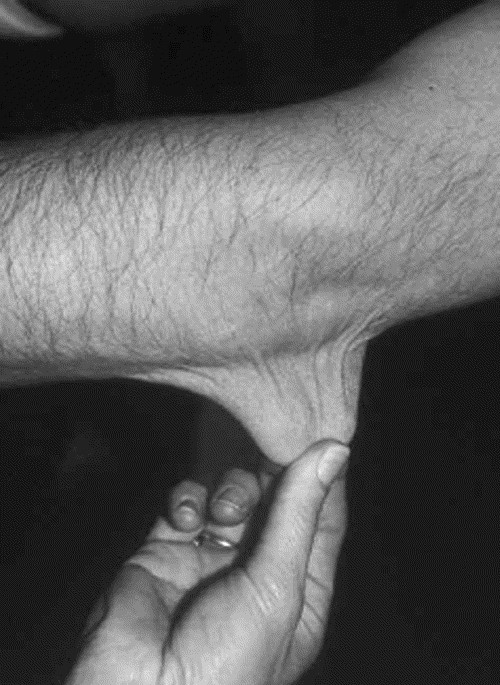
Skin hyperextensibility in a patient with EDS, classic type.
-
Widened atrophic scarring (a manifestation of tissue fragility) (Fig. 2).
Fig. 2 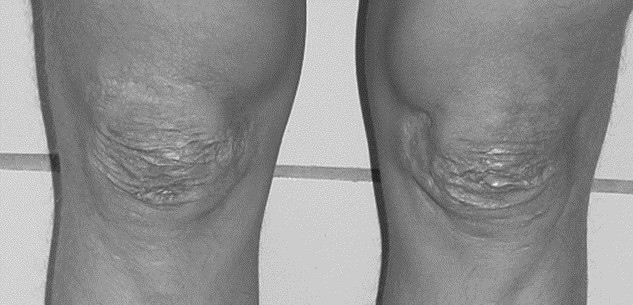
Widened atrophic scars on the knees in a patient with EDS, classic type.
-
Joint hypermobility: Joint hypermobility depends on age, gender, and family and ethnic backgrounds. Joint hypermobility in classic EDS is generalized, affecting both large and small joints and can range in severity from mild to severe. It is usually noted when a child starts to walk. It should be assessed using the Beighton scale,3 the most widely accepted grading system for the objective semiquantification of joint hypermobility (Table 1).
Table 1 Beighton criteria for joint hypermobility -
Positive family history.


Minor diagnostic criteria were also established, and presence of one or more of these minor criteria contributes to the diagnosis of classic EDS but is not sufficient to establish the diagnosis:
-
Smooth, velvety skin.
-
Molluscoid pseudotumors (fleshy, heaped-up lesions associated with scars over pressure points such as the elbows and knees).
-
Subcutaneous spheroids (small, hard cyst-like nodules, freely moveable in the subcutis over the bony prominences of the legs and arms, which have an outer calcified layer with a translucent core on x-ray).
-
Complications of joint hypermobility (e.g., sprains, dislocations/subluxations, and pes planus).
-
Muscle hypotonia, delayed gross motor development.
-
Easy bruising.
-
Manifestations of tissue extensibility and fragility (e.g., hiatal hernia, anal prolapse in childhood, and cervical insufficiency).
-
Surgical complications (postoperative hernias).
Cutaneous manifestations
Cutaneous hyperextensibility (Fig. 1) is one of the cardinal features of EDS in general and of classic EDS in particular. Skin extends easily and snaps back after release (unlike the lax and redundant skin observed in the cutis laxa syndromes). The skin is typically very smooth and velvety to the touch. It is also very fragile, as manifested by splitting of the dermis following relatively minor trauma, especially over pressure points (elbows and knees), and areas prone to trauma (forehead, chin, shins). Skin fragility may cause dehiscence of sutured incisions in skin or mucosa. Wounds take a longer time to heal and stretching of scars after apparently successful primary wound healing is characteristic. Scars become wide, with a “cigarette-paper”-like or papyraceous appearance (Fig. 2). Other dermatologic features in classic EDS include molluscoid pseudotumors, subcutaneous spheroids, and piezogenic papules (small, painful, reversible herniations of underlying adipose tissue globules through the fascia into the dermis, such as on medial and lateral aspects of the feet on standing). Less frequent cutaneous manifestations include acrocyanosis (a painless disorder caused by constriction or narrowing of the small blood vessels in the skin, mainly the hands in which the affected areas turn blue and become cold and sweaty, and localized swelling may also occur) and chilblains (cold injuries, characterized by a red swollen skin that is tender, hot to the touch and may itch; it can develop in less than 2 hours in skin exposed to cold). Elastosis perforans serpiginosa, a skin condition of unknown etiology characterized by skin colored to erythematous keratotic papules, some enlarging outward in serpiginous or arcuate configurations, leaving slightly atrophic centers, rarely occurs.
Musculoskeletal manifestations
Joint hypermobility may lead to major articular complications, such as habitual subluxation and dislocation of the joints most often affecting shoulder, patella, digits, hip, radius, and clavicles. These dislocations may occur and usually resolve spontaneously or are easily managed by the patient. Some individuals with classic EDS may suffer from chronic joint and limb pain, despite normal skeletal radiographs. Other problems related to the joint hypermobility are joint instability, foot deformities such as congenital clubfoot or pes planus, temporomandibular joint dysfunction, joint effusions, and osteoarthritis. At birth, uni- or bilateral dislocation of the hip may be present.4–6
Easy bruising
Easy bruising is a common finding and manifests as spontaneous ecchymoses, frequently recurring in the same areas and causing a characteristic brownish discoloration of the skin, especially in exposed areas such as shins and knees. In small children, easy bruising may be the presenting symptom to the pediatrician. There is a tendency toward prolonged bleeding, e.g., following brushing of the teeth, despite a normal coagulation status.
This bleeding diathesis is explained by an abnormal capillary structure with deficiency of normal perivascular collagen, resulting in poor support of cutaneous blood vessels, which rupture when subjected to shearing forces. Laboratory investigation of platelet aggregation, clotting factors, and bleeding time in patients with EDS is usually normal. The Rumpel-Leede (or Hess) test may be positive, indicating capillary fragility.7,8
Manifestations of tissue fragility
Manifestations of generalized tissue extensibility and fragility may be observed in multiple organs. Patients with classic EDS often suffer from repetitive hernia, such as inguinal, umbilical, hiatal, or incisional hernia. In early childhood, recurrent rectal prolapse may be observed.
Facial features
Patients with classic EDS display typical facial characteristics such as epicanthic folds, excess skin on the eyelids, some dilated scars on the forehead and the chin, and a pale and sometimes a somewhat prematurely aged aspect of the facies.
Neurologic features
Primary muscular hypotonia may occur and may cause delayed motor development, problems with ambulation, and mild motor disturbance. Fatigue and muscle cramps are relatively frequent. Cerebral spine fluid leak has rarely been reported to cause postural hypotension and headache in individuals with classic EDS.9
Cardiovascular manifestations
Structural cardiac malformations are uncommon in the classic type of EDS. Mitral valve prolapse and, less frequently, tricuspid valve prolapse may occur. Stringent criteria should be used for the diagnosis of mitral valve prolapse. Aortic root dilatation may be more common than previously thought.10 Spontaneous rupture of large arteries, along with intracranial aneurysms and arteriovenous fistulae, may occur in the rare individual with a severe form of classic EDS but is more frequently associated with vascular EDS.
Pregnancy-related manifestations
Pregnancy in a woman with classic EDS bears risk for the newborn and for the woman. Especially in the more severe form of classic EDS, prematurity occurs more often if the fetus is affected, mostly due to premature rupture of the membranes. Because of hypotonia, breech presentation is more frequent if the baby is affected and may lead to dislocation of the hips or shoulder of the newborn. For an affected woman, there is also an increased risk for extension of episiotomy incisions, tearing of the perineal skin, and prolapse of the uterus and/or the bladder may occur after delivery. As a whole, these complications are more frequent than in the normal population. However, it is difficult to quantitate the incidence of each complication in affected individuals, as no good studies exist.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis with other EDS-subtypes
Other forms of EDS should be considered in individuals with easy bruising, joint hypermobility, and/or chronic joint dislocation. The disorders in which clinical findings overlap with the classic type of EDS include the following.
EDS hypermobility type (EDS type III)
In this form, joint hypermobility is the primary manifestation. The skin is often soft or velvety and may be mildly hyperextensible. Subluxations and dislocations are common; they may occur spontaneously or with minimal trauma and can be acutely painful. Degenerative joint disease is common. Chronic pain, distinct from that associated with acute dislocations or advanced osteoarthritis, is a serious complication of the condition and can be both physically and psychologically disabling. Easy bruising is common. Joint hypermobility is the primary clinical manifestation. Skin abnormalities, such as variable skin hyperextensibility and smooth velvety skin, are found, but the presence of large atrophic scars in individuals with joint hypermobility suggests the diagnosis of classic EDS.
The diagnosis of EDS, hypermobility type is based entirely on clinical evaluation and family history. In most individuals with EDS, hypermobility type, the causative gene is unknown and unmapped.11 Haploinsufficiency of TNXB and heterozygosity for missense mutations in TNXB, the gene encoding tenascin X, have been associated with EDS, hypermobility type in a small subset of affected individuals.12,13 A single occurrence of a COL3A1 mutation in a family thought to have EDS, hypermobility type has been reported.14 Inheritance is autosomal dominant.
Familial joint hypermobility syndrome
Familial joint hypermobility syndrome and other syndromes in which joint hypermobility is found, share hypermobility of the joints with classic EDS, but the absence of skin hyperextensibility and atrophic scarring excludes the diagnosis of classic EDS.
Tenascin X deficiency
Homozygous mutations have been identified in TNXB in a few individuals with an autosomal-recessive EDS phenotype characterized by joint hypermobility, skin hyperextensibility without atrophic scarring, easy bruising and occasionally increased laxity of the genitourinary tract causing uterine and vaginal prolapse, and increased risk for postpartum hemorrhage.15–17 Heterozygotes for the same mutation, especially females, appear to have an EDS hypermobility phenotype.
EDS vascular type (EDS type IV)
This condition is characterized by thin, translucent skin, easy bruising, and a characteristic facial appearance. Most importantly, however, affected individuals are at risk for arterial rupture, aneurysm, or dissection; gastrointestinal perforation or rupture; and uterine rupture during pregnancy. One fourth of individuals with EDS vascular type experience a significant medical problem by age 20 years and more than 80% by age 40 years. The mean age of death is 48 years.18 This autosomal condition is caused by mutations in the COL3A1 gene, encoding the α1-chain of type III collagen. The diagnosis is based on clinical findings and confirmed by biochemical and/or molecular genetic testing. Biochemical studies demonstrate abnormal electrophoretic mobility and/or abnormal efficiency of secretion of type III procollagen by cultured dermal fibroblasts. Molecular genetic testing is used to identify mutations in the COL3A1 gene.
EDS kyphoscoliotic type (EDS type VI)
EDS kyphoscoliotic type is a generalized connective tissue disorder characterized by kyphoscoliosis, joint laxity, muscle hypotonia, and, in some individuals, fragility of the ocular globe. Tissue fragility (including atrophic scars) and skin hyperextensibility are usually present. Affected individuals are at risk for rupture of medium sized arteries and respiratory compromise if kyphoscoliosis is severe. EDS kyphoscoliotic type is caused by deficient activity of the enzyme procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD1: lysyl hydroxylase 1). The diagnosis of EDS kyphoscoliotic form relies on the demonstration of an increased ratio of deoxypyridinoline to pyridinoline crosslinks in urine measured by HPLC, a highly sensitive and specific test. Assay of lysyl hydroxylases enzyme activity in skin fibroblasts is also available. Molecular genetic testing of the PLOD1 gene is available. Inheritance is autosomal recessive.
EDS arthrochalasia type (EDS type VIIA and B)
EDS arthrochalasia type is an autosomal-dominant disorder, distinguished by severe joint hypermobility at birth and congenital bilateral hip dislocation. Tissue fragility (including atrophic scars) and skin hyperextensibility are usually present; severity ranges from mild to severe. It is caused by mutations in COL1A1 or COL1A2 leading to skipping of all or part of exon 6 of the messenger RNA (mRNA) coding for one of the α1 chains (EDS VIIA) or the α2 chain (EDS VIIB) of type I collagen, respectively.
EDS dermatosparaxis type (EDS type VIIC)
This autosomal-recessive condition is characterized by extreme skin fragility and skin laxity, but the skin has a sagging, redundant appearance. Other distinctive features are delayed closure of the fontanels, characteristic facies, edema of the eyelids, blue sclerae, umbilical hernia, short fingers, and short stature. The disorder is caused by deficient activity of procollagen-N-proteinase, the enzyme that excises the amino (N)-terminal propeptide in procollagen types I, II, and III.19–21
EDS progeroid form
This is a rare autosomal-recessive disorder characterized by progeroid appearance with wrinkled facies, curly and fine hair, scanty eyebrows and eye lashes, and periodontitis, in addition to typical signs of EDS. It is caused by homozygous mutations in β4GALT7, the gene encoding galactosyltransferase I. This enzyme catalyzes the second glycosyl-transfer reaction in the assembly of the dermatan sulfate chain.
EDS cardiac valvular form
The cardiac valvular form of EDS is characterized by joint hypermobility, skin hyperextensibility with variable atrophic scarring, and cardiac valvular defects. Total absence of the proα2(I) chains of type I collagen as a result of homozygous or compound heterozygous mutations in the COL1A2 gene is causative.22,23 Inheritance is autosomal recessive.
Classic-like EDS with propensity for arterial rupture
One arginine-to-cysteine (R-to-C) substitution in proα1(I) chain of type I collagen (R134C) has been identified in a series of patients with a condition reminiscent of classic EDS, with skin hyperextensibility, easy bruising, atrophic scarring, and joint hypermobility but with propensity for arterial rupture at adult age.24,25 Two other proα1(I) R-to-C substitutions (R396C and R915C) were also associated with rupture of medium sized arteries, but affected individuals did not present EDS-like skin features.25 Furthermore, a proα1(I)-R888C substitution was reported in a family presenting an EDS/osteogenesis imperfecta (OI) overlap phenotype,26 and a proα1(I)-R836C was shown to be associated with autosomal-dominant Caffey disease.27
Periventricular heterotopia, EDS variant
Periventricular heterotopia belongs to a clinically and genetically heterogeneous group of neuronal migration disorders. Human Filamin A gene mutations are associated with classical X-linked bilateral periventricular nodular heterotopia.
Recently, atypical phenotypes, including bilateral periventricular nodular heterotopia with EDS have been described in a couple of patients.28–34 EDS features include joint hypermobility, skin hyperextensibility with variable presence of widened atrophic scarring, and development of aortic dilatation in early adulthood.
EDS/OI overlap phenotype
Some defects in the most amino-terminal region of either the α1(I)- or the α2(I)-collagen triple helix have been shown to result in a EDS/OI overlap phenotype, characterized on the one hand by joint hypermobility, skin hyperexentibility with mildly atrophic scarring, and easy bruising and on the other hand by a variable degree of bone fragility, short stature, and blue sclerae.35 Some patients also suffered from severe bleeding problems, suggesting vascular fragility (Unpublished data).
Occipital horn syndrome (formerly known as EDS type IX)
Occipital horn syndrome (OHS) is characterized by “occipital horns,” distinctive wedge-shaped calcifications at the sites of attachment of the trapezius muscle and the sternocleidomastoid muscle to the occipital bone. Occipital horns may be clinically palpable or observed on skull radiographs. Individuals with OHS also have lax skin and joints, bladder diverticula, inguinal hernias, and vascular tortuosity. There is no particular easy bruising or fragility of the skin. Serum copper concentration and serum ceruloplasmin concentrations are low. A multiplex protocol of targeted mutation analysis, mutation scanning, and sequence analysis detects mutations in ATP7A in more than 95% of affected individuals.36 Inheritance is X linked.
Newly recognized rare EDS variants
Recently, two new autosomal-recessive EDS variants were recognized, which show some overlap with classic EDS but which can readily be distinguished by other clinical features.
The spondylocheirodyplastic form of EDS is characterized by hyperextensible thin skin, easy bruising, hypermobility of the small joints with a tendency to contractures, protuberant eyes with bluish sclerae, hands with finely wrinkled palms, atrophy of the thenar muscles, and tapering fingers. Skeletal surveys show platyspondyly with moderate short stature, osteopenia, and widened metaphyses. The condition is caused by mutations in the SLC39A13 gene, encoding the membrane-bound zinc transporter SLC39A13.37
The RIN2-syndrome is characterized by severe progressive scoliosis, progressive facial coarsening, gingival hypertrophy, sparse hair, and skin and joint hyperlaxity. It is caused by mutations in the RIN2 gene. RIN2 encodes the Ras and Rab interactor-2, which acts as a guanine nucleotide exchange factor for the small GTPase Rab5, which is involved in early endocytosis.38,39
Differential diagnosis with other heritable connective tissue disorders
Classic EDS shows limited overlap with other connective tissue disorders, including variants of the following, but these disorders are differentiated by other distinctive clinical features.
Marfan syndrome
Marfan syndrome has a broad continuum of clinical manifestations involving the ocular, skeletal, and cardiovascular systems. Lens dislocation, seen in about 60%, is a hallmark feature. Myopia, retinal detachment, glaucoma, and early cataract formation are seen. Bone overgrowth leads to long extremities and pectus deformity (excavatum or carinatum) and joint laxity; scoliosis is common. Cardiovascular manifestations include dilatation of the aorta, a predisposition for aortic tear and rupture, mitral valve prolapse with or without regurgitation, tricuspid valve prolapse, and enlargement of the proximal pulmonary artery. Marfan syndrome is a clinical diagnosis based on family history and the observation of characteristic findings in multiple organ systems. Diagnostic criteria have been established. Molecular genetic testing of the FBN1 gene is possible. Inheritance is autosomal dominant.
Loeys-Dietz syndrome
Loeys-Dietz syndrome is an aortic aneurysm syndrome characterized by a triad of hypertelorism, bifid uvula/cleft palate, and arterial tortuosity with ascending aortic aneurysm/dissection. The main differences with Marfan syndrome are the absence of long bone overgrowth and lens dislocations, and the presence of multiple other findings, including craniosynostosis, Chiari malformation, club feet, patent ductus arteriosus, and aneurysms and dissections throughout the arterial tree. In contrast to this typical presentation, which was later coined as Loeys-Dietz syndrome type I, some patients show less craniofacial abnormalities but more prominent joint and skin manifestations, reminiscent of EDS. This subset of patients (referred to as Loeys-Dietz-syndrome type II) is characterized by easy bruising, a velvety translucent skin, widened atrophic scars, uterine rupture, severe peripartal bleeding, and arterial aneurysm/dissection throughout the arterial circulation.
The natural history of Loeys-Dietz syndrome types I and II is far more aggressive than Marfan syndrome or even vascular EDS, with a mean age of death of 26 years. Aortic dissections occur in young childhood and/or at smaller aortic dimensions, and the incidence of pregnancy-related complications is high.40 The condition is caused by heterozygous mutations in the TGFBR1 and TGFBR2 genes, encoding the transforming growth factor-beta receptors 1 and 2. Inheritance is autosomal dominant.
Cutis laxa syndromes
Hyperextensible skin should also be distinguished from that observed in the cutis laxa syndromes and in De Barsy syndrome, in which the redundant skin hangs in loose folds and only returns very slowly to its former position. In these syndromes, the skin is not fragile, and wound healing is normal. The cutis laxa syndromes result from the loss or fragmentation of the elastic fiber network. They are variably associated with pulmonary, cardiac, arterial, and gastrointestinal abnormalities. Cutis laxa syndromes comprise autosomal-dominant, autosomal- recessive, and X-linked forms. The autosomal-dominant form is caused by mutations in the ELN gene, encoding elastin. Autosomal-recessive forms of cutis laxa are associated with mutations in the genes encoding fibulin 4 and fibulin 5 (FBLN4 and FBLN5),41,42 and more recently also with mutations in ATP6V0A2 and PYCR1.43,44 The previously defined X-linked form of cutis laxa, or OHS, is caused by mutations in ATP7A and was discussed earlier.
MANAGEMENT
Initial evaluation
We recommend the following examinations to establish the extent of disease in an individual with classic EDS at time of diagnosis.
-
Good clinical inspection of the skin with assessment of skin hyperextensibility, atrophic scars and bruises, and other manifestations of classic EDS.
-
Evaluation of joint mobility with use of the Beighton score.
-
Evaluation for hypotonia and motor development in infants and children.
-
A baseline echocardiogram with aortic diameter measurement for those younger than 10 years.
-
Evaluation of clotting factors if severe easy bruising is present.
Treatment of manifestations
Cutaneous manifestations
Dermal wounds should be closed with tension, preferably in two layers, and deep stitches should be applied generously. Cutaneous stitches should be left in place twice as long as usual, and additional fixation of adjacent skin with adhesive tape can help prevent stretching of the scar.
Musculoskeletal manifestations
In children with hypotonia and delayed motor development, a physiotherapeutic program is important. Non weight-bearing muscular exercise, such as swimming, is useful to promote muscular development and coordination. Antiinflammatory drugs may help with joint pain. Surgical stabilization of joints may lead to disappointing, or only temporary, improvement.
Prevention of primary manifestations
Cutaneous manifestations and bruising
Patients with EDS should be instructed to avoid undue trauma to the skin and other organ systems. Children with pronounced skin fragility should wear protective pads or bandages over the forehead, knees, and shins from an early age.
Patients with pronounced bruising should avoid contact sports and heavy exercise. Protective pads and bandages are also useful in the prevention of bruises and hematomas. Supplementation of ascorbic acid (Vitamin C), a cofactor for crosslinking of collagen fibrils, ameliorates the tendency toward bruising in some patients.45 Ascorbic acid (Vitamin C) may reduce easy bruising but has no effect on the primary findings of skin hyperextensibility, atrophic scarring, and joint hypermobility. In general, 2 g per day is recommended for adults, with proportionally reduced doses for children; however, there is no limitation. No strict recommendations exist regarding the third trimester dose. Desamino-d-arginine-vasopressin may be useful in patients with EDS with chronic bruising or epistaxis, or perioperatively (e.g., for tooth extraction), in whom bleeding time is normalized by desamino-d-argininevasopressin.46,47
Musculoskeletal manifestations
Excessive stretching of the joints should be avoided as this further exacerbates the underlying disorder. Children should be refrained from “showing off” by demonstrating their joint laxity to others. Individuals with muscle hypotonia and joint instability with chronic pain may have to adjust lifestyle and professional choices accordingly. They should avoid excessive or repetitive heavy lifting and other movements that produce undue strain on the already hypermobile joints.
Surveillance
Cardiovascular manifestations
If no abnormalities are found on echocardiogram in an adult, a follow-up echocardiogram is not necessary. Because longitudinal data on progression of aortic dilation are not available, specific recommendations for follow-up in individuals with a normal aortic diameter are not available. If an abnormality such as aortic dilatation or mitral valve prolapse is found on echocardiogram, follow-up echocardiogram should be performed once a year. Cardiovascular problems should be treated in a standard manner.
Pregnancy
Careful follow-up is recommended throughout pregnancy and postpartum. Monitoring is warranted during the third trimester when the risk of premature rupture of the membranes is increased.
Emotional support
Emotional support and behavioral and psychological therapy may be indicated to accept and cope with the handicap and the long-term chronic pain. Patient support groups are available and can be very beneficial.
LABORATORY TESTING
Ultrastructural studies
Electron microscopy of a skin biopsy in EDS, classic type often suggests disturbed collagen fibrillogenesis. A “cauliflower” deformity of collagen fibrils is characteristic.48 However, these findings are not specific for EDS and, therefore, not diagnostic. Furthermore, ultrastructural changes, usually most pronounced in the central parts of the reticular dermis, may be missed if the skin biopsy is not of full thickness.
Biochemical testing
Collagen protein analysis is performed on cultured fibroblasts, derived from a skin biopsy to obtain a source of protein for electrophoretic analysis of collagen types I, III, and V. The collagens are labeled and analyzed on sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Abnormal proteins will migrate differently on the gel, when compared with control samples.49 However, because type V collagen is synthesized by fibroblasts at low levels, alterations in electrophoretic mobility are poorly reproducible, making this an ineffective method for routine diagnostic evaluation. The test, however, helps to exclude other subtypes of EDS, such as the vascular, kyphoscoliotic, arthrochalasis, and the dermatosparaxis type in individuals in which clinical differential diagnosis is difficult. Rarely, an abnormal electrophoretic pattern for type I collagen is detected because of a arginine-tocysteine substitution in the COL1A1 gene coding for the proα1(I) collagen chain of type I collagen.24,25
Molecular testing
Studies suggest that approximately 50% of individuals with classic EDS have an identifiable mutation in the COL5A1 or COL5A2 gene, the genes encoding type V collagen. However, because no prospective molecular studies of COL5A1 and COL5A2 have been performed in a clinically well-defined patient group, this number may underestimate the real proportion of patients with classic EDS harboring a mutation in one of these genes.
Molecular investigations usually start from genomic DNA (gDNA) and mRNA, extracted from cultured dermal fibroblasts. As a first step, a COL5A1 “null-”allele test is performed, using polymorphic markers in the expressed region of the gDNA, to determine whether both COL5A1 alleles have stable transcripts. This test determines whether the individual is heterozygous for one of several COL5A1 polymorphic exonic markers in gDNA and then establishes at the complementary (cDNA) level whether both alleles are expressed. If only one of the two COL5A1 alleles is present in cDNA, it is assumed that the absent allele is “null.” Because it examines both gDNA and cDNA, COL5A1 “null” allele testing requires cultured skin fibroblasts. It does not identify mutations within the COL5A1 gene. COL5A1 null alleles are detected in approximately 30–40% of individuals with classic EDS.49
Sequence analysis by Sanger sequencing of the COL5A1 and the COL5A2 gene can be performed either on gDNA or cDNA. Once the mutation is known, its presence or absence can easily be verified on gDNA obtained from leukocytes from the patient and from relatives. Most families harbor a unique mutation in either COL5A1 or COL5A2.
Linkage analysis can be offered to patients with a positive family history for classic EDS. These studies are based on the accurate clinical diagnosis in the affected family members, and they are dependent on the willingness and the availability of family members to be tested, and the presence of informative polymorphic markers.
MOLECULAR PATHOGENESIS
Type V collagen is a quantitatively minor fibrillar collagen, which is present in much smaller amounts than other fibril-forming collagens, but it is widely distributed in a variety of tissues such as skin, tendon, bone, cornea, placenta, and fetal membranes. It occurs as two forms of heterotrimers ([α1(V)]2α2(V) and α1(V)α2(V)α3(V)) or as α1(V)3 homotrimers. In most vertebrate tissues, such as skin, tendon, and bone, it is present mainly as [α1(V)]2α2(V) heterotrimers. The α1(V)3 homotrimers may exist in normal tissues, whereas the α1(V)α2(V)α3(V) heterotrimers are present mainly in placenta. Type V collagen coassembles with type I collagen to form heterotypic fibrils. The entire triple helical domain of type V collagen is buried within the heterotypic fibril, and only the retained amino-terminal (NH2) propeptide of type V collagen is exposed at the surface. This NH2-propeptide has a regulatory function in the process of fibril assembly, and it regulates the diameter of the heterotypic fibrils.50 Recent data indicate that type V collagen controls collagen fibril assembly in several tissues.51COL5A1 haploinsufficiency in a murine model for classic EDS was shown to result in abnormal fibril nucleation and dysfunctional fibril growth with potential disruption of cell directed fibril organization.52
The COL5A1 gene, located at 9q34.2-q34.3, is a large gene, comprising 66 exons distributed over >150 kb of gDNA, and it encodes the α1 chain of type V collagen. The COL5A2 gene, located at 2q31, comprises 52 exons distributed over 67 kb and encodes the α2 chain of type V collagen. The α3 chain of type V collagen, encoded by the COL5A3 gene, is located on chromosome 19p13.2.
The most common types of molecular defect lead to haploinsufficiency for the COL5A1 mRNA. In approximately one third of individuals with classic EDS, nonsense or frameshift mutations are responsible for a nonfunctional COL5A1 allele.49,53–55 Nonsense, frameshift, or splice site mutations that introduce a premature termination codon are usually responsible for this nonfunctional COL5A1 allele. A variety of mechanisms lead to nonsense-mediated decay of the mutation-bearing mRNA. The predicted consequence is synthesis of about half the amount of normal type V collagen.
Structural mutations in the COL5A1, which exert a dominant-negative effect, have been demonstrated in a few individuals with classic EDS. In a small proportion of individuals, a mutation affects the structural integrity of type V collagen, resulting in the production of a functionally defective type V collagen protein (dominant-negative mutation). These structural mutations are most commonly splice site mutations that result in exon skipping and a few point mutations that result in the substitution for glycine in the triple-helical region of the collagen molecule.49,55–61 In contrast to other disorders characterized by mutations in the fibrillar collagen genes, remarkably few mutations have been found that result from the substitution of a glycine by a bulkier amino acid.
Two COL5A1 mutations have been reported deleting one or more highly conserved cysteine residues in the C-terminal propeptide of the α1(V) collagen chain, which are essential for intrachain disulphide bonding before assembly of the proα-chains and initiation of triple-helix folding of fibrillar collagen molecules. Most likely, these mutations prevent incorporation of the mutant proα1(V)-chains into the molecule and chain degradation. This seems to be another mechanism for haploinsufficiency but at the protein level.57,60
More recently, two leucine substitutions in the signal peptide domain of the preproα1(V)-chain of type V collagen were reported to cause classic EDS. We showed that these substitutions impair secretion of mutant type V collagen protein to the extracellular environment, suggesting interference with intracellular protein trafficking to the endoplasmic reticulum.62 Taken together, these findings imply a similar mechanism of action for type V collagen SP mutations as for C-propeptide mutations, or mutations resulting in a nonfunctional COL5A1 allele, in that they all result in a reduced amount of normal type V collagen available for collagen fibrillogenesis.
A G530S substitution located in the amino-terminal propeptide of the α1(V) chain was suggested to be disease modifying when present in the heterozygous state and disease causing in the homozygous state.61,63 At present, however, its contribution to the pathogenesis of classic EDS remains unclear.55
PENETRANCE AND GENOTYPE-PHENOTYPE CORRELATIONS
The number of patients described with mutations in COL5A1 or COL5A2 is relatively small. Inter- and intrafamilial variability in the severity of the phenotype can be great. No distinct genotype-phenotype correlations have emerged so far. In particular, no difference in severity is noted between patients with mutations resulting in nonsense-mediated decay of one COL5A1 allele, when compared with patients with a structural mutation in COL5A1 or COL5A2, or in patients in whom no mutation is detected. Intrafamilial variability in severity can be strikingly large. This is illustrated by two pedigrees with classic EDS, where the affected children had a “full-blown” classic EDS phenotype, fulfilling the major diagnostic criteria, whereas the affected parent only showed some mild and localized joint hypermobility and no overt skin hyperextensibility or scarring. Molecular testing showed the presence of a nonfunctional COL5A1 allele in the affected children and the affected parent in both pedigrees. These examples illustrate the large phenotypic variability of classic EDS, even in one family, and the possibility of the presence of a COL5A1 null allele in individuals with only minor symptoms of classic EDS. They also show that there exists a clinical overlap between mild classic EDS (the former type II) and the hypermobility type of EDS.
GENETIC COUNSELING
EDS, classic type is inherited in an autosomal-dominant manner. It is estimated that approximately 50% of affected individuals have inherited the mutant gene from an affected parent, and about 50% of affected individuals have a de novo disease-causing mutation. The parents of a proband with an apparent de novo mutation should be evaluated by physical examination of the skin with special attention to delayed wound healing and abnormal scarring, easy bruising, joint hypermobility or recurrent dislocations, and chronic articular pain. Although about 50% of individuals diagnosed with classic EDS have an affected parent, the family history may seem to be negative because of failure to recognize the disorder in family members.
The risk to sibs of the proband depends on whether one of the proband's parents has classic EDS. If a parent is affected, the risk to the sibs is 50%. When the parents are clinically unaffected, the risk to the sibs of a proband seems to be low. Although no instances of germline mosaicism have been reported, it remains a theoretical possibility in a minority of cases.
Each child of an individual with classic EDS has a 50% chance of inheriting the mutation. The risk to other family members depends on the status of the proband's parents. If a parent is found to be affected, his/her family members are at risk.
The optimal time for determination of genetic risk and genetic counseling regarding prenatal testing is before pregnancy. No laboratories offering molecular genetic testing for prenatal diagnosis for classic EDS are listed in the GeneTests Laboratory Directory. However, prenatal testing may be available for families in which linkage has been established or the disease-causing mutation has been identified in an affected family member in a research or clinical laboratory.
Requests for prenatal testing for conditions such as classic EDS that do not affect intellect or life span are not common. Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. Although most centers would consider decisions about prenatal testing to be the choice of the parents, careful discussion of these issues is appropriate.
Preimplantation genetic diagnosis may be available for families in which the disease-causing mutation has been identified in an affected family member in a research or clinical laboratory.
References
Byers P Disorders of collagen biosynthesis and structure. In: Scriver CR, Beaudet AR, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited d isease, 2nd ed. Edinburgh, Churchill Livingstone, 2001; 1065–1081.
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ . Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet 1998; 77: 31–37.
Beighton P, de Paepe A, Danks D, et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am J Med Genet 1988; 29: 581–594.
Hagberg C, Berglund B, Korpe L, Andersson-Norinder J . Ehlers-Danlos syndrome (EDS) focusing on oral symptoms: a questionnaire study. Orthod Craniofac Res 2004; 7: 178–185.
De Coster PJ, Martens LC, De Paepe A . Oral health in prevalent types of Ehlers-Danlos syndromes. J Oral Pathol Med 2005; 34: 298–307.
De Coster PJ, Van den Berghe LI, Martens LC . Generalized joint hypermobility and temporomandibular disorders: inherited connective tissue disease as a model with maximum expression. J Orofac Pain 2005; 19: 47–57.
De Paepe A, Malfait F . Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. Br J Haematol 2004; 127: 491–500.
Malfait F, De Paepe A . Bleeding in the heritable connective tissue disorders: mechanisms, diagnosis and treatment. Blood Rev 2009; 23: 191–197.
Schievink WI, Gordon OK, Tourje J Connective tissue disorders with spontaneous spinal cerebrospinal fluid leaks and intracranial hypotension: a prospective study. Neurosurgery 2004; 54: 65–70; discussion 70–61.
Wenstrup RJ, Meyer RA, Lyle JS, et al. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genet Med 2002; 4: 112–117.
Malfait F, Hakim AJ, De Paepe A, Grahame R . The genetic basis of the joint hypermobility syndromes. Rheumatology 2006; 45: 502–507.
Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet 2003; 73: 214–217.
Zweers MC, Dean WB, van Kuppevelt TH, Bristow J, Schalkwijk J . Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin Genet 2005; 67: 330–334.
Narcisi P, Richards AJ, Ferguson SD, Pope FM . A family with Ehlers-Danlos syndrome type III/articular hypermobility syndrome has a glycine 637 to serine substitution in type III collagen. Hum Mol Genet 1994; 3: 1617–1620.
Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med 2001; 345: 1167–1175.
Lindor NM, Bristow J . Tenascin-X deficiency in autosomal recessive Ehlers-Danlos syndrome. Am J Med Genet A 2005; 135: 75–80.
Egging DF, van Vlijmen-Willems I, Choi J, et al. Analysis of obstetric complications and uterine connective tissue in tenascin-X-deficient humans and mice. Cell Tissue Res 2008; 332: 523–532.
Pepin M, Schwarze U, Superti-Furga A, Byers PH . Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000; 342: 673–680.
Colige A, Sieron AL, Li SW, et al. Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am J Hum Genet 1999; 65: 308–317.
Colige A, Ruggiero F, Vandenberghe I, et al. Domains and maturation processes that regulate the activity of ADAMTS-2, a metalloproteinase cleaving the aminopropeptide of fibrillar procollagens types I-III and V. J Biol Chem 2005; 280: 34397–34408.
Malfait F, De Coster P, Hausser I, et al. The natural history, including orofacial features of three patients with Ehlers-Danlos syndrome, dermatosparaxis type (EDS type VIIC). Am J Med Genet A 2004; 131: 18–28.
Schwarze U, Hata R, McKusick VA, Shinkai H, Hoyme HE, Pyeritz RE, et al. Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Hum Genet 2004; 74: 917–930.
Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A . Total absence of the alpha2(I) chain of collagen type I is a rare cause of Ehlers-Danlos Syndrome hypermobility and propensity to cardiac valvular problems. J Med Genet 2006; 43: e36.
Nuytinck L, Freund M, Lagae L, Pierard GE, Hermanns-Le T, De Paepe A . Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen. Am J Hum Genet 2000; 66: 1398–1402.
Malfait F, Symoens S, De Backer J, et al. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat 2007; 28: 387–395.
Cabral WA, Makareeva E, Letocha AD, et al. Y-position cysteine substitution in type I collagen (alpha1(I) R888C/p.R1066C) is associated with osteogenesis imperfecta/Ehlers-Danlos syndrome phenotype. Hum Mutat 2007; 28: 396–405.
Gensure RC, Makitie O, Barclay C, et al. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest 2005; 115: 1250–1257.
Sole G, Coupry I, Rooryck C, et al. Bilateral periventricular nodular heterotopia in France: frequency of mutations in FLNA, phenotypic heterogeneity and spectrum of mutations. J Neurol Neurosurg Psychiatry 2009; 80: 1394–1398.
Savasta S, Crispino M, Valli M, Calligaro A, Zambelloni C, Poggiani C . Subependymal periventricular heterotopias in a patient with Ehlers-Danlos syndrome: a new case. J Child Neurol 2007; 22: 317–320.
Parrini E, Ramazzotti A, Dobyns WB, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain 2006; 129: 1892–1906.
Sheen VL, Walsh CA . Periventricular heterotopia: new insights into Ehlers-Danlos syndrome. Clin Med Res 2005; 3: 229–233.
Gomez-Garre P, Seijo M, Gutierrez-Delicado E, et al. Ehlers-Danlos syndrome and periventricular nodular heterotopia in a Spanish family with a single FLNA mutation. J Med Genet 2006; 43: 232–237.
Sheen VL, Jansen A, Chen MH, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology 2005; 64: 254–262.
Thomas P, Bossan A, Lacour JP, Chanalet S, Ortonne JP, Chatel M . Ehlers-Danlos syndrome with subependymal periventricular heterotopias. Neurology 1996; 46: 1165–1167.
Cabral WA, Makareeva E, Colige A, et al. Mutations near amino end of alpha 1(I) collagen cause combined OI/EDS by interference with N-propeptide processing. J Biol Chem 2005; 280: 19259–19269.
Kaler SG ATP7A-related copper transport disorders. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [internet]. Seattle, Washington: University of Washington, Seattle, 1993–2003. Updated July 13, 2005.
Giunta C, Elcioglu NH, Albrecht B, et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome—an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am J Hum Genet 2008; 82: 1290–1305.
Basel-Vanagaite L, Sarig O, Hershkovitz D, et al. RIN2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: MACS syndrome. Am J Hum Genet 2009; 85: 254–263.
Syx D, Malfait F, Van Laer L, et al. The RIN2 syndrome: a new autosomal recessive connective tissue disorder caused by deficiency of Ras and Rab interactor 2 (RIN2). Hum Genet 2010; 128: 79–88.
Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 24 2006; 355: 788–798.
Hucthagowder V, Sausgruber N, Kim KH, Angle B, Marmorstein LY, Urban Z . Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am J Hum Genet 2006; 78: 1075–1080.
Loeys B, Van Maldergem L, Mortier G, et al. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet 2002; 11: 2113–2118.
Kornak U, Reynders E, Dimopoulou A, et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet 2008; 40: 32–34.
Reversade B, Escande-Beillard N, Dimopoulou A, et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet 2009; 41: 1016–1021.
Steinmann B, Royce P, Superti-Furga A The Ehlers-Danlos syndrome. In: Royce P, Steinmann B (eds) Connective tissue and its heritable disorders. 2nd ed. New York, Wiley-Liss, Inc, 2002; 431–523.
Stine KC, Becton DL . DDAVP therapy controls bleeding in Ehlers-Danlos syndrome. J Pediatr Hematol Oncol 1997; 19: 156–158.
Mast KJ, Nunes ME, Ruymann FB, Kerlin BA . Desmopressin responsiveness in children with Ehlers-Danlos syndrome associated bleeding symptoms. Br J Haematol 2009; 144: 230–233.
Hausser I, Anton-Lamprecht I . Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet 1994; 93: 394–407.
Malfait F, Coucke P, Symoens S, Loeys B, Nuytinck L, De Paepe A . The molecular basis of classic Ehlers-Danlos syndrome: a comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum Mutat 2005; 25: 28–37.
Birk DE . Type V collagen: heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron 2001; 32: 223–237.
Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE . Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem 2004; 279: 53331–53337.
Wenstrup RJ, Florer JB, Davidson JM, et al. Murine model of the Ehlers-Danlos syndrome. col5a1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J Biol Chem 2006; 281: 12888–12895.
Wenstrup RJ, Florer JB, Willing MC, et al. COL5A1 haploinsufficiency is a common molecular mechanism underlying the classical form of EDS. Am J Hum Genet 2000; 66: 1766–1776.
Schwarze U, Atkinson M, Hoffman GG, Greenspan DS, Byers PH . Null alleles of the COL5A1 gene of type V collagen are a cause of the classical forms of Ehlers-Danlos syndrome (types I and II). Am J Hum Genet 2000; 66: 1757–1765.
Mitchell AL, Schwarze U, Jennings JF, Byers PH . Molecular mechanisms of classical Ehlers-Danlos syndrome (EDS). Human Mutat 2009; 30: 995–1002.
Nicholls AC, Oliver JE, McCarron S, Harrison JB, Greenspan DS, Pope FM . An exon skipping mutation of a type V collagen gene (COL5A1) in Ehlers-Danlos syndrome. J Med Genet 1996; 33: 940–946.
Wenstrup RJ, Langland GT, Willing MC, D'Souza VN, Cole WG . A splice-junction mutation in the region of COL5A1 that codes for the carboxyl propeptide of pro alpha 1(V) chains results in the gravis form of the Ehlers-Danlos syndrome (type I). Hum Mol Genet 1996; 5: 1733–1736.
Michalickova K, Susic M, Willing MC, Wenstrup RJ, Cole WG . Mutations of the alpha2(V) chain of type V collagen impair matrix assembly and produce Ehlers-Danlos syndrome type I. Hum Mol Genet 1998; 7: 249–255.
Burrows NP, Nicholls AC, Richards AJ, et al. A point mutation in an intronic branch site results in aberrant splicing of COL5A1 and in Ehlers-Danlos syndrome type II in two British families. Am J Hum Genet 1998; 63: 390–398.
De Paepe A, Nuytinck L, Hausser I, Anton-Lamprecht I, Naeyaert JM . Mutations in the COL5A1 gene are causal in the Ehlers-Danlos syndromes I and II. Am J Hum Genet 1997; 60: 547–554.
Giunta C, Steinmann B . Compound heterozygosity for a disease-causing G1489E [correction of G1489D] and disease-modifying G530S substitution in COL5A1 of a patient with the classical type of Ehlers-Danlos syndrome: an explanation of intrafamilial variability?. Am J Med Genet 2000; 90: 72–79.
Symoens S, Malfait F, Renard M, et al. COL5A1 signal peptide mutations interfere with protein secretion and cause classic Ehlers-Danlos syndrome. Human Mutat 2009; 30: E395–E403.
Giunta C, Nuytinck L, Raghunath M, Hausser I, De Paepe A, Steinmann B . Homozygous Gly530Ser substitution in COL5A1 causes mild classical Ehlers-Danlos syndrome. Am J Med Genet 2002; 109: 284–290.
Acknowledgements
This work was supported by a Methusalem Grant BOF08/01M01108 from the Flemish Government and the Ghent University (to A.D.P.). Fransiska Malfait is a postdoctoral research fellow paid from the fund for scientific research, Flanders.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Malfait, F., Wenstrup, R. & De Paepe, A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet Med 12, 597–605 (2010). https://doi.org/10.1097/GIM.0b013e3181eed412
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181eed412
Keywords
This article is cited by
-
Psychological interventions for individuals with Ehlers-Danlos syndrome and hypermobility spectrum disorder: a scoping review
Orphanet Journal of Rare Diseases (2023)
-
Classic Physical Exam Findings in Ehlers-Danlos Syndrome
Journal of General Internal Medicine (2023)
-
Thoracoabdominal aortic aneurysm in connective tissue disorder patients
Indian Journal of Thoracic and Cardiovascular Surgery (2022)
-
Biomechanical properties of the patellar tendon in children with heritable connective tissue disorders
European Journal of Applied Physiology (2018)
-
COL5A1 gene variants previously associated with reduced soft tissue injury risk are associated with elite athlete status in rugby
BMC Genomics (2017)
