
Abstract
This review is in two sections. The first section summarises 35 conditions, both common and infrequent, causing cicatrising conjunctivitis. Guidelines for making a diagnosis are given together with the use of diagnostic tests, including direct and indirect immunofluorescence, and their interpretation. The second section evaluates our knowledge of ocular mucous membrane pemphigoid, which is the commonest cause of cicatrizing conjunctivitis in most developed countries. The clinical characteristics, demographics, and clinical signs of the disease are described. This is followed by a review and re-evaluation of the pathogenesis of conjunctival inflammation in mucous membrane pemphigoid (MMP), resulting in a revised hypothesis of the autoimmune mechanisms causing inflammation in ocular MMP. The relationship between inflammation and scarring in MMP conjunctiva is described. Recent research, describing the role of aldehyde dehydrogenase (ALDH) and retinoic acid (RA) in both the initiation and perpetuation of profibrotic activity in MMP conjunctival fibroblasts is summarised and the potential for antifibrotic therapy, using ALDH inhibition, is discussed. The importance of the management of the ocular surface in MMP is briefly summarised. This is followed with the rationale for the use of systemic immunomodulatory therapy, currently the standard of care for patients with active ocular MMP. The evidence for the use of these drugs is summarised and guidelines given for their use. Finally, the areas for research and innovation in the next decade are reviewed including the need for better diagnostics, markers of disease activity, and the potential for biological and topical therapies for both inflammation and scarring.
Similar content being viewed by others

Introduction
It is a great privilege to have been asked to give this lecture and I am very grateful to the Scientific Committee of the College for inviting me to undertake it. I am going to discuss the results of 25 years of studies both collaborating with, and supported by, a large number of colleagues, including scientists and clinicians and patients. I will use our studies in the context of the available evidence base to describe the causes and diagnosis of cicatrising conjunctivitis (CC) and follow this with a review of mucous membrane pemphigoid (MMP), which is the most common cause in developed countries. I hope that what I have to say about a group of diseases that is challenging for both patients and clinicians, will interest, entertain and challenge you as much as it has me.
All previous Bowman Lecturers have felt that their predecessors have been hard acts to follow and I am no different. However, all of us have worked in the shadow of Sir William Bowman. The portrait in Figure 1 has rarely been seen. It was painted by George Watts in about 1865 when Bowman was at the height of his powers. Watts was a friend of Bowman’s and the leading portraitist of the day. Like many others who have given this lecture I became fascinated by Bowman’s life and achievements. I wanted to know what it was that made him one of the foremost scientists of his day for his work on histology, bringing him Fellowship of the Royal Society at the age of 25, and then going on to become one of the founders of Ophthalmology as a scientific discipline in the UK. I have drawn on the biographies by his contemporary Power,1 and by James2 for the background, but also on other sources to make some connections that have not, to my knowledge, been made before. These have been summarised in Supplementary Appendix 1.

Portrait of Sir William Bowman by George Watts painted about 1865. I am grateful to Rachel Clarkson, Sir William’s great-great-granddaughter, for providing this image.
In the early 19th century the classification of conjunctivitis was almost unrecognizable from what we know today. However, by the end of the 19th century trachoma, trauma, ocular rosacea, adenovirus, allergic eye disease, lupus and pemphigus (at that time a term including all the pemphigoid diseases) had been recognized. A brief history of milestones in the recognition of the diseases causing CC is summarised in Supplementary Appendix 2 and Supplementary Table 1.
Current causes, epidemiology and classification of cicatrising conjunctivitis
Table 1 lists causes of CC from two studies. Thorne (2004)3 provides the largest institutional case series of CC cases attending an MMP clinic, either to have the cause of CC diagnosed, or to have ocular involvement by MMP excluded from those patients with an established diagnosis of extraocular MMP. But, causes in single clinics are skewed by case selection and referral bias. For this reason, the epidemiology of CC is best described by the study from Radford (2012)4 in the UK, which lists the causes of cicatrizing conjunctivitis from the only national incidence study to have been reported. This study may have underestimated the incidence of CC by 15%, but the figures for MMP are in line with those reported for that disease from French and German estimates of 1.13 per million in France and 0.87 per million in Germany.4
Table 2 lists 30 causes of CC with references and notes about the associated diseases and the rarity of the conditions. Although not complete, it includes those reported causes for which there is reasonable evidence of causation. These diseases have been divided into three categories in Table 2, each of which relate to current treatment protocols.
Blinding scarring diseases
Blinding scarring diseases include mucous membrane pemphigoid (MMP), a rare subset of cases of drug induced scarring, and Stevens-Johnson syndrome/Toxic epidermal necrolysis (SJS/TEN), that behave like MMP and develop both inflammation and progressive scarring.
Ocular surface neoplasia
Ocular surface neoplasia (OSN) is an uncommon cause of inflammation and scarring which may mimic MMP although it is usually, but not always, unilateral unlike MMP which is usually, but not always, bilateral. The oncology protocols required for treatment will be delayed by misdiagnosis as CC.
Other conjunctival scarring diseases
Other conjunctival scarring diseases are those that have frequently been termed ‘pseudopemphigoid’. The reason for this distinction, and for the term pseudopemphigoid, has been summarized in the study from Thorne et al3 comparing MMP and with pseudopemphigoid3 where pseudopemphigoid is used as the term for non MMP causes of CC. ‘Other ocular surface diseases can cause cicatrizing conjunctivitis and symblepharon formation that mimic MMP. These diseases may be grouped under the term 'pseudopemphigoid'. Although there are uncommon diseases associated with pseudopemphigoid that might require systemic immunosuppressive drug therapy (e.g., lichen planus or paraneoplastic pemphigus), most of the causes of pseudopemphigoid do not require systemic immunosuppressive drug therapy. Because long-term, systemic immune-suppressive drug therapy has potentially life-threatening side effects, differentiating ocular MMP from pseudopemphigoid is essential for the proper treatment of patients with cicatrizing conjunctivitis’.
However, the use of ‘pseudopemphigoid’ as a catch-all term for any cause of CC that shares the clinical features of MMP, but which demands a different therapeutic approach, is too non-specific to be helpful for several reasons. It groups diseases with very different aetiologies, some of which may be associated with severe inflammation, unresponsive to topical therapy, that require immunosuppressive treatment. These include severe atopic keratoconjunctivitis (AKC),5 graft versus host disease (GVHD),6 sarcoid,7 ectrodactyly-ectodermal dysplasia-cleft syndrome EEC syndrome, and ectodermal dysplasia (ED),8 even though the associated scarring rarely leads to pathology. In addition, some of these conditions, with an established diagnosis, such as AKC and lupus, may also have concomitant MMP, which must be excluded. For these reasons the term pseudopemphigoid, as a specific term covering a disparate group of diseases, can be confusing and is best avoided.
Making the diagnosis
The diagnostic problem is shown by the similarity of the clinical appearances of the diseases illustrated in Figure 2. This has resulted in the difficulty we have in distinguishing many of the causes in Table 2 from ocular MMP and making a firm diagnosis, particularly for eye diseases without systemic associations or with dual pathology such as SJS/TEN, and for CC triggered by eye drops. Figure 3 summarises these dilemmas. Despite the similarities in the clinical characteristics some features of the clinical examination can help, as can a carefully taken history, and the appropriate investigations providing their limitations are understood.

Illustrations of some of the causes of cicatrising conjunctivitis (a) Mucous membrane pemphigoid, (b) Trachoma, (c) Adenovirus, (d) Sebaceous carcinoma, (e) Ocular surface squamous neoplasia, (f) Ectrodactyly Ectodermal Dysplasia Cleft Lip/Palate (g) Glaucoma drops, (h) Atopic keratoconjunctivitis, (i) Stevens-Johnson syndrome, (j) Ocular rosacea, (k) Lichen Planus, (l) Sarcoid.

The diagnostic problem in cicatrising conjunctivitis. This Figure illustrates the classification of CC used in Table 2 with notes on the prevalence, which causes are often overlooked, which are usually known but which may co-exist with mucous membrane pemphigoid and causes associated with systemic diseases that may have their first manifestations in the eye.
Clinical examination
Given the similarity of the clinical phenotype, clinical examination is principally useful for establishing whether the disease is unilateral, and therefore potentially caused by ocular surface neoplasia, and to identify signs associated with a few of the other causes. Ocular MMP is rarely unilateral occurring in only 7/115 (6%) of cases in one of our series.9 Other clinical findings that contribute are: severe aqueous tear deficiency (rare in early ocular MMP) caused by both Sjögren’s syndrome and severe non-Sjögren’s dry eye, for which a low Schirmer’s 1 test (without anaesthesia) is indicative although not diagnostic because of limited sensitivity.10 For ocular rosacea the scarring is associated with meibomitis and a papillary and follicular conjunctivitis with or without the following: pseudopterygium, peripheral corneal vascularisation and usually the dermatological features of acne rosacea.11
History
The other disorders listed in Table 2 have a clear history of an associated ocular condition or systemic disease with the exceptions listed below. Direct questions about the eye diseases, risks associated with scarring, and systemic diseases that are associated with CC (listed in Table 2) will identify whether the patient has an ocular or systemic disease in association with CC. This information will not always be volunteered by patients or referring physicians. The diseases which develop CC, without a history of an associated systemic disease, or which present in the eye before disease develops elsewhere are:
-
Ocular MMP (conjunctival disease without involvement of other sites) has comprised 32–48% of 132 cases in 2 UK studies including both cases with and without identified autoantibodies but meeting clinical criteria.4, 12
-
Paraneoplastic pemphigoid and pemphigus (PNP) may rarely present in the conjunctiva at the same time as a neoplasm, and before other signs of malignancy.13
-
Pemphigus vulgaris (PV) may present in the conjunctiva before other signs: one case series of this presentation has been described.14
-
Sarcoid may present in the conjunctiva and be associated with severe scarring.7 Conjunctival biopsy, even in the absence of conjunctival signs, can be diagnostic in 30% of patients with systemic sarcoidosis.15
The rate of progression of scarring can usually be established by taking a careful history of the onset of symptoms of inflammation and scarring. The scarring may have been noticed by a healthcare professional or as a result of the onset of trichiasis. Estimating the rate of progression is important when deciding on the need for therapy, given the diagnostic delay for patients with ocular MMP (mean of 2.5 years and a range up to 10 years).4
Patients with multiple causes of CC
As described in Table 2 patients with SJS/TEN and drug induced CC may rarely develop disease that is indistinguishable from ocular MMP. We also have one unreported case of MMP who later developed localised conjunctival OSSN. Other causes of CC including trachoma and atopic keratoconjunctivitis may also develop MMP and this can be expected to occur occasionally for any of the other causes. It follows that any patient with rapid progression of scarring and inflammation should have investigations to exclude MMP and OSSN.
When to investigate?
Unilateral cases (to exclude OSN), patients without a clear history of an associated ocular or systemic disease presenting in the eye (such as occurs in ocular MMP, PNP, PV, and Sarcoid), and those with an ocular or systemic disease, but with inflammation and scarring not responding to topical therapy. The latter is most common in SJS/TEN, and in topical drug associated CC, for whom there is often reluctance to discontinue topical therapy for advanced glaucoma. In the latter group severe inflammation and scarring may be associated with most non preserved glaucoma medications, as well as due to preservatives:16 the inflammation will often rapidly resolve on withdrawal of the drops although this may take 2–6 weeks to improve and longer to resolve.17 Oral acetazolamide will often be tolerated by this group of patients, at least for 1–2 weeks to confirm the diagnosis of toxicity/allergy to topical medications. If oral acetazolamide is not tolerated, topical unpreserved iopidine, to which few of these patients have been exposed, can be used as an alternative. We have had to carry out glaucoma tube surgery on some patients having these reactions.
Which investigations to use?
Our protocol for the investigation of CC cases is included in Supplementary Appendix 3.
Blood tests
Blood tests are used in our department for the following:
-
a
Indirect immunofluorescence and individual autoantibody detection to identify specific autoantibodies associated with MMP and PV.
Indirect immunofluorescence
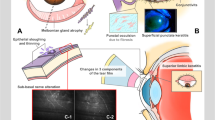
Indirect immunofluorescence (IIF) microscopy on salt-split skin is used to identify the presence of serum autoantibodies in pemphigoid diseases. The structure of the basement membrane, its constituent proteins and the identification of these by immunofluorescence tests is illustrated in Figures 4 and 5.18 However, IIF is non-specific for individual antibodies which cannot be identified from this test. The technique is clearly described by Hintner.19

The epithelial basement membrane structure and immunofluorescent tests. The cartoon describes the basement membrane (dermoepidermal junction) structure and its constituent proteins. The proteins that have been shown to be target antigens in MMP are listed. The position of the basement membrane (BM) is shown on a direct immunofluorescence (DIF) specimen which also shows positive immunofluorescence to IgG in the BM zone. An example of indirect immunofluorescence (IIF) on salt split skin is also shown demonstrating positive fluorescence to the floor (see Figure 5). The cartoon is reproduced with permission from Figure 1 Schmidt.18

Autoimmune bullous dermatoses and lichen planus. Direct immunofluorescence (DIF) and indirect immunofluorescence. The illustrations in the top left panel shows pemphigus vulgaris (PV) with DIF on perilesional skin showing intraepithelial antibody binding. In the top right panel the skin lesions in bullous pemphigoid are shown for comparison with those in PV together with an example of a positive DIF in conjunctiva with antibody binding at the basement membrane. The pemphigus and pemphigoid diseases are listed separating those that may be associated with conjunctival scarring and those that are not. The panel on mucosal lichen planus shows the typical appearance of a ‘shaggy’ fibrin staining band at the dermoepidermal junction. The bottom panel shows indirect immunofluorescence on human salt split skin from a patient with MMP.
Autoantibody detection
Autoantibodies to BP180, laminin 332 BP230, α6β4 integrin, laminin 311, type VII collagen, Laminin γ1 (p200 protein) are associated with MMP and those to the intraepithelial proteins desmoglein 1 and 3 are associated with pemphigus vulgaris,20 which may occasionally present in the eye. ELISAs for antibodies to BP180, BP230, Collagen VII, Desmoglein 1, and Desmoglein 3 are available from immunodermatology services and some laboratories offer immunoblotting to detect the remaining autoantibodies.
Although often negative in MMP, positive indirect immunofluorescence, or autoantibody detection, showing circulating anti-basement membrane zone antibodies provides additional evidence for an underlying autoimmune pathology21 and may occasionally be positive when DIF is negative.3
-
a
Autoantibody screening, and inflammatory markers to identify patients at risk of subclinical autoimmune disease which is present in over 30% patients with MMP (compared with less than 10% in a control population).22
-
b
Baseline screening for patients who might need immunosuppressive therapy: full blood count, blood film, glucose 6 phosphate dehydrogenase (G6PD) level (dapsone is contraindicated if this is elevated, which is common in Mediterranean races), thiopurine methyltransferase (TPMT) levels relevant to metabolism of azathioprine, urea and electrolytes, creatinine, liver function tests, glucose and infection markers including quantiferon gold (for TB), hepatitis and HIV.
Biopsies
Biopsies are taken for routine histopathology and direct immunofluorescence studies. Bulbar conjunctival biopsies are safe to take in patients with probable or proven MMP: of 344 cases having bulbar conjunctival biopsy no adverse effects were reported.23, 24, 25 However, Foster reported that fornix conjunctival biopsies may be associated with an exacerbation of scarring in 3 cases, following which he abandoned this site for diagnostic biopsy.23 The technique for taking biopsies has recently been described.26
-
a
Routine histopathology with fixation in 10% formol saline is used to identify atopic keratoconjunctivitis,27 sarcoid15 and is essential to exclude OSN, particularly in unilateral CC.
-
b
Direct immunofluorescence (DIF) from perilesional tissue is required to confirm the diagnosis in the immunobullous diseases.19, 28 Figure 5 illustrates the differences between the DIF findings for the intraepithelial ‘pemphigus’ diseases and the subepithelial 'pemphigoid' diseases, first defined by Lever in 1953.29 Lichen planus, an inflammatory disease affecting skin and sometimes the oral and ocular mucosa, is also shown. DIF is used to differentiate these conditions and is described in Figure 5.
Interpretation of DIF results
Causes of cicatrising conjunctivitis which may be DIF positive from the conjunctiva and skin, or other mucosal sites, include
-
MMP (including mucosal dominated EB. Linear IgA disease and anti-laminin 332 pemphigoid), some patients with SJS/TEN, some with drug-induced scarring, all of which show IgG or IgA and/or complement on DIF at the epithelial basement membrane (shown in Figures 4 and 5), should be treated as MMP cases. Note that for the diagnosis of ocular MMP a conjunctival biopsy is NOT required for the diagnosis if there has been a positive biopsy from any other site (skin, buccal, genital, and nasopharyngeal mucosa), providing the ocular findings are typical of MMP.21 The DIF findings in BP are the same as for MMP so that the diseases must be differentiated on clinical grounds.
-
Pemphigus vulgaris may rarely present in the conjunctiva as conjunctivitis in addition to conjunctival scarring, and conjunctival DIF has shown the characteristic epithelial intercellular fluorescence to IgG and/or complement14 shown in Figure 5, that is typically found in the skin at the dermoepidermal junction (basement membrane zone) in PV.19
-
Lichen planus has a characteristic 'ragged' fibrin basement membrane deposition as shown in Figure 5.21
Other diseases associated with CC, and which may have positive conjunctival DIF are
-
The ectodermal dysplasias with a pattern identical to that of MMP in the conjunctiva.8
-
Lupus shows granular deposition of IgG, IgA, IgM, and complement at the dermal basement membrane in many cases19 and also in the conjunctiva in CC cases associated with lupus.30
Conjunctival scarring associated disease which may be DIF positive from skin, but not from the conjunctiva
-
Conjunctival DIF has not been demonstrated in paraneoplastic pemphigus, which may also be associated with conjunctival scarring, and show a similar pattern to that of PV, in addition to a pemphigoid like pattern at the dermal epidermal junction.
-
Dermatitis herpetiformis having a typical granular pattern at the basement membrane zone.19
-
Porphyria cutanea tarda also shows characteristic immunoglobulin deposition at the dermoepidermal junction but one study of associated conjunctival scarring showed repeatedly negative DIF results.31
Negative immunofluorescence findings in ocular MMP
There is a strong recommendation, from an influential consensus document,21 that MMP can only be diagnosed when both clinical criteria and direct immunopathology criteria have been fulfilled. The clinical criteria are those distinguishing MMP from other diseases involving mucous membranes. Those that are listed in the document, and which cause CC, are lichen planus, SJS/TEN, PV, PNP, BP, and drug-induced CC, to which the additional disorders summarized in Table 2 must be added. The direct immunopathology criteria that are recommended, as mandatory for diagnosis, are either DIF microscopy, which is widely available, or immunohistochemistry which is not. Both positive IIF or specific autoantibody detection are widely accepted as alternative evidence when DIF findings are negative.
These criteria are usually positive in MMP that involves tissues other than the eye. However negative DIF results have long been recognized as presenting a diagnostic problem in patients with strong clinical evidence of ocular only MMP (in whom the conjunctiva is the only site of involvement). In three studies describing this group of 49 patients3, 24, 32 25/49 (51%) were DIF+ of whom 13 (26%) required more than one biopsy to demonstrate this (Supplementary Table 2). Other problems with the use of DIF as a mandatory finding for the diagnosis of MMP are that the results can be initially positive, then subsequently negative when patients are in remission.24 Identical biopsy findings are found in bullous pemphigoid, which has to be distinguished by the clinical findings, and identical findings are also reported in some patients with SJS/TEN,33 drug-induced progressive scarring,34 ectodermal dysplasia,8 and ulcerative colitis.32
Immunopathology, as described in the consensus document, refers to the autoantibody detection tests (DIF, IIF or other means of autoantibody detection) that are recommended for diagnosis in these immunobullous disorders. However, the presence of autoantibodies does not reflect the dominance of the cell mediated autoreactive response, to mucosal basement membrane epitopes, which is probably important in many cases of ocular MMP (see sections Loss of tolerance...in MMP and Autoantibodies in normal controls in the review of the pathogenesis of ocular MMP below). In effect a positive DIF result, as recommended for diagnosis, is a biomarker for an autoantibody driven autoimmune pathology and not the cellular autoimmune response. Unfortunately, DIF has not proved to be either sensitive in ocular MMP, nor specific as a marker for ocular MMP. Although a positive DIF result is useful, and can distinguish MMP from lichen planus, lupus erythematosus and PV and PNP, which have characteristic immune-pathological features of their own, a negative result does not exclude ocular MMP. Currently patients are being seen at Moorfields who could benefit from treatment for MMP, but who have been untreated because of uncertainty about the diagnosis due to their having negative autoantibody results. This has resulted in delayed therapy and deterioration of their scarring and inflammation. For these reasons we have proposed new criteria for the diagnosis of ocular MMP that are summarised below.
Recommended diagnostic criteria for ocular MMP
We have proposed three sets of criteria for the diagnosis of ocular MMP:4, 35, 36
-
a
Patients with positive conjunctival DIF or positive DIF from another site meet currently agreed criteria.21
-
b
Patients with negative DIF from any site and positive indirect immunofluorescence can be diagnosed as having MMP.
-
c
Patients with negative immunopathology can be diagnosed with ocular MMP providing that they have a typical phenotype of progressive conjunctival scarring, and that other diseases that may cause this phenotype have been excluded. When ocular cases are reported, the detailed immunopathology findings should be recorded so that the diagnosis can be interpreted in light of future modifications to diagnostic criteria.
When to refer?
Patients with scarring and inflammation if there are no facilities for the specialist investigations or for delivering immunomodulatory therapies, for rare diseases, when the diagnosis is uncertain and when there is a poor response to therapy. In these circumstances a specialist centre with expertise in CC can provide the resources both to clarify the diagnosis and establish a treatment plan.4 Guidelines for referral are given in Figure 6.

Review of mucous membrane pemphigoid with ocular involvement
This is the commonest disease causing cicatrising conjunctivitis in the United Kingdom, accounting for 60% of CC cases, and is the prototypic autoimmune mucosal scarring disease. For the latter reasons MMP has been the focus of much of the research into scarring eye diseases in developed countries and ocular involvement by MMP has threefold more citations on PubMed than any other cause of CC apart from trachoma. MMP has been the focus of most of my work in CC and is the topic of the rest of this review.
Terminology
The terminology for the immunobullous diseases is confusing, given the changes in nomenclature over the past 250 years, and more recently. All these bullous diseases were categorized as pemphigus, until Lever’s seminal paper in which he differentiated the intraepithelial pemphigus diseases, from the subepithelial pemphigoid (meaning ‘resembling pemphigus’) diseases. This separation of these diseases was based on his histological studies demonstrating the tissue that was the focus of the inflammatory response. Since then the immunobullous diseases have been classified into these two groups, which are described in Figure 5.
The terminology for MMP has also changed. Initially Lever’s term for this was Benign Mucous Membrane Pemphigoid (BMMP) because, unlike pemphigus vulgaris, which had a very high mortality before the introduction of steroids, it was rarely fatal. In the 1980’s the term BMMP was replaced by the term cicatricial pemphigoid (CP) to reflect the associated scarring. CP and MMP have been used interchangeably at different times, and the studies describing the sites of involvement have prefaced these terms with the name of the site, most often ocular CP (OCP) and ocular MMP (OMMP). Cicatricial pemphigoid is a term that is now used to describe a rare disease, having the same immunopathology as MMP, but which is limited to the head and neck and is also called localised CP or Brunsting Perry cicatricial pemphigoid.37 The term OCP is still in occasional use although, following the 2002 Consensus document21 the term MMP has been recommended for any patient, with any site of involvement (because multiple sites are often involved). However, this recommendation is impractical for specialist publications, describing the diagnosis and management of the different mucosal sites, and the descriptive terms ocular MMP (instead of OCP) and oral MMP are now being used for studies focused on diseases at these sites.
The other principal terminological difficulty is the altering nomenclature of the basement membrane immunoreactant proteins: examples are BP180, also known as Bullous Pemphigoid Antigen II and classified as Collagen type XVII, BP230 is also known as Bullous Pemphigoid Antigen I, laminin 332, formerly called laminin 5 or epiligrin, and p200 protein which is now designated as laminin γ1.
Clinical characteristics
Sites of involvement Mucous membrane pemphigoid involves all the orificial mucosal sites (oral, ocular, nasopharyngeal, genital and anal) as well as, less often, tracheal and oesophageal. The skin may also be involved. Figure 7 shows examples of disease at many of the sites involved by MMP. All mucosal sites may become severely inflamed however scarring is rare in the oral mucosa whereas scarring is mandatory for the diagnosis in the conjunctiva. Oral MMP, when restricted to the oral mucosa, has a relatively benign course in many patients, unlike ocular MMP. The conjunctiva is involved in 70% of all MMP cases resulting in bilateral blindness in 20% of cases and severe sight loss in 30% of eyes.9, 12, 38 In a cross sectional study (unpublished) between 2009–2012 of 73 MMP patients with conjunctival involvement seen at Moorfields, 27% of patients had ocular only MMP, 26% ocular and oral, and a further 26% ocular, oral and nasopharyngeal, with the remainder having a mixture of other sites involved.

The spectrum of disease in mucous membrane pemphigoid (MMP) (a) Gingival inflammation and ulceration, (b) Palatal inflammation and ulceration, (c) Supraglottic inflammation and scarring, (d) Oesophageal stenosis, (e) Skin ulceration and scarring, (f) Foreskin scarring, (g) Conjunctival inflammation and scarring, (h) Intraoperative photograph showing a normal subconjunctival space under incised severely scarred and shortened conjunctiva demonstrating that subconjunctival tissue is unaffected.
Demographics In the UK national incidence survey which included 50 MMP patients meeting clinical criteria for the disease the median age was 71 years (range 20-90), there was a preponderance of males with a M:F ratio of 29:21 (1.38:1), with 32/50 (64%) having extraocular MMP (as opposed to ocular only MMP) of whom 8 (16%) had another autoimmune disease and 26/40 (65%) were taking immunosuppressive therapy at a 12 month follow up.4 These finding are similar to those in an institutional case series of 50 patients with a median age of 67 years (range 32–91), M:F ratio of 23:27 (0.85:1), extraocular MMP in 26/50 (52%) and 38/50 (76%) requiring immunosuppressive therapy after their initial assessment at the specialist centres.12 There is no good data on racial predisposition although MMP is probably less common in Indian Asians39 and the Chinese40, 41 than in Caucasians. The disease is reported in Japan.
Clinical signs of disease progression
Early diagnosis and initiation of appropriate treatment are essential to prevent the sight-threatening complications of the ~75% of ocular MMP patients with rapidly progressive disease.42 Clinical signs are illustrated in Figure 8, ocular MMP typically presents with a red eye and persistent conjunctivitis that has not responded to topical therapy, or with cicatricial entropion and trichiasis, that may have failed surgical repair. About 30% of patients present with acute conjunctivitis and limbitis leading to rapidly progressive scarring and surface failure if uncontrolled.42 Similarly, persistent epithelial defect has a poor prognosis occurring in about 20% of patients. The remaining patients present with subacute or low grade chronic inflammation and slowly progressive scarring. The earliest clinical sign in patients with subacute disease is often medial canthal scarring, with loss of the plica and caruncle. Medial canthal scarring is usually an early sign of MMP and is not as frequent in conjunctival scarring due to other causes. Linear scarring in the sulcus subtarsalis (marginal sulcus) of the upper tarsus is sometimes present early in the disease. Other signs, in order of progression, are subepithelial reticular fibrosis, infiltration of the tarsal and bulbar43, 44 conjunctiva, shortening of the fornices, symblepharon and cicatricial entropion, followed by ankyloblepharon and then, subsequent to scarring of the lacrimal ductules which usually occurs late in the disease, a totally dry ‘skin like’ eye. Figure 9 describes the events leading to morbidity and blindness in ocular MMP.

Ocular mucous membrane pemphigoid (MMP). Clinical signs in ocular MMP.

Factors contributing to progression of disease in ocular mucous membrane pemphigoid (MMP).
Pathogenesis
Predisposing factors
As described in the section on the demographics of MMP above the disease is probably more common in Caucasians than in Indian and Chinese Asians, although it may occur in any racial group. Also, other autoimmune diseases are more common in MMP patients. Many patients have a genetic predisposition to MMP, expressing the HLA-DQB*0301 gene.45 For the majority of patients there are no identifiable precipitating factors. However, in subsets of patients, including cases of ocular MMP following SJS/TEN33, 46 and topical glaucoma treatment,34, 47 it is possible that damage to the conjunctival basement membrane precipitates the disease by exposing basement membrane epitopes triggering a pathological autoimmune response to neoantigens. The latter mechanism is an alternative to the development of loss of tolerance to basement membrane antigens, that is described below, and thought to be the underlying mechanisms in most cases.
Loss of tolerance, autoantibodies and autoreactive T cells and mechanisms of disease activation and remission at different sites in MMP
As in other autoimmune disorders, disease probably develops as a result of loss of tolerance. In the case of the autoimmune subepithelial bullous dermatoses, of which MMP is one, this loss of tolerance is to epithelial basement membrane proteins. This has been shown to result in circulating autoreactive T cells in two cases48 and in the generation of autoantibodies to a number of basement membrane proteins, described in Figure 4most commonly BP 180. The pathogenic potential of antibodies has been demonstrated: anti-laminin 332 induces blistering in a mouse model and anti-α6β4 integrin induces separation of the dermoepidermal junction in organ culture of human skin.18 In serum, antibody levels have been correlated with disease activity in MMP.49 As a result of these findings it has been proposed that autoantibodies must be demonstrated for the diagnosis of MMP21 as for the other pemphigoid diseases.18
On the other hand, not all patients with MMP have demonstrable circulating autoantibodies. In ocular MMP only 50% of patients have these (see section on negative immunofluorescence findings in ocular MMP, above). In addition, few MMP patients suffer from involvement of all potential sites of involvement: of 112 patients with MMP in a cross sectional study in London (unpublished) 2/112 patients had 5 involved sites, and 6/112 had 4 involved sites, from a potential maximum of 7 sites (ocular, oral, skin, nasopharyngeal, laryngeal, genital, and perianal). The mechanisms that protect individual sites from involvement in a systemic disease, caused by circulating antibodies to proteins common to the basement membranes of all the target tissues, may involve factors local both to those tissues and to the local cell mediated inflammatory response. The latter is a feature of these diseases18, 50 in which autoreactive T cells are central both to the autoantibody response, and to the cellular autoimmune response.48 Combined cellular and antibody mediated responses are common in autoimmune diseases, all of which require autoreactive T cells. Although some autoimmune diseases are dominated by the pathogenic effects of one effector pathway, either autoantibodies or effector T cells, both pathways are commonly involved. Examples are Graves’ disease which is an autoantibody dominated disease, mediated by autoantibodies to thyroid stimulating hormone. At the other end of the spectrum, psoriasis is the result of an autoreactive T cell dominated response to skin associated antigens. However, most autoimmune diseases, exemplified by systemic lupus erythematosus and rheumatoid arthritis, result from the effects of both autoantibody and autoreactive T cell mediated inflammation in the target tissues.51 In bullous pemphigoid there is ample evidence for combined autoimmune cellular and antibody mediated effector pathways,18 as there is for mucous membrane pemphigoid.52
Autoimmune reactivity is normal in healthy individuals who may express both autoantibodies and autoreactive T cells without developing an effector response that results in disease.51 In the last decade naturally occurring autoantibodies have been shown to have homeostatic functions in the clearance of oxidatively damaged body waste, and in the modulation of immune cell functions.53
Autoantibodies in normal MMP controls
Several studies have examined the prevalence of autoantibodies to BP180 and BP230, the basement membrane proteins commonly precipitating an autoreactive response in pemphigoid diseases and show that these are probably present in about 10-15% of healthy age matched individuals. However, the prevalence of these antibodies has varied in relation to the sensitivity and specificity of the assay used and the control group. The lowest prevalence has been less than 1% in a large cohort of blood donors of unknown ages,54 other publications have given values of 7.5%,55 13%, 16% and 26% for ELISA and or immunoblots56 although higher values have been reported.57 Results for IIF have generally shown <5% positivity55, 56 although this was 19% (n=32) in another study.58 Autoreactive T cells recognising the basement membrane protein BP 180, are also present in healthy individuals expressing the HLA-DQB*0301 gene (although not in those who did not).59 These autoreactive T cells are thought to be prevented from developing a pathological response by the activity of both regulatory T cells (Treg)60 and regulatory B cells (Breg).61 The role of these regulatory cells has been evaluated in only two studies in MMP: Treg were shown to be present in higher numbers in MMP lesional tissue compared with the skin of normal controls52 whereas a study of peripheral blood in patients, with a variety of pemphigus and pemphigoid diseases, showed reduced numbers of cells with Breg characteristics, compared with controls, in pemphigus only and not in pemphigoid.62
In summary, a substantial proportion of normal individuals have antibodies to both BP180 and BP230, some also having autoreactive T cells to BP180. There is evidence that Treg are present in MMP lesional tissue. We can hypothesise that differences in the balance between autoreactive T cells and Tregs in the different lesional MMP tissues account for the development and resolution of the disease in different target tissues in any one individual affected by MMP. If this occurs disease activation and remission in MMP is likely to be occurring at a local lesional level in the tissues, and independent of circulating autoantibody production and local antibody deposition. If this hypothesis is correct then the balance, between autoantibody and autoreactive cellular effector mechanisms, may differ both for different sites, and at different stages of disease chronicity, accounting for the lack of evidence of circulating antibodies in some cases of MMP, particularly in those with ocular MMP.
However, this is an area which demands further study, and the control mechanisms are likely to be more complex than this. Given that anti-CD20 B cell depletion therapy (such as the anti-CD20 monoclonal, rituximab) is effective in many cases of T-cell-mediated diseases such as type I diabetes and rheumatoid arthritis, as well as MMP, the role of B cells in regulating T-cell responses has been examined63 and evidence suggests that, in some circumstances, B cell antigen presentation to autoreactive T cells may be necessary to develop an autoreactive T-cell response. In addition, unstimulated B cells may also promote the development of Treg cells.63
Summary of the pathogenesis of inflammation and scarring in ocular MMP
This is described in Figure 10, for which the evidence is summarised both in Table 3 ‘Effectors and cytokines identified in MMP conjunctiva’ and in the hypothesis synopsised above, that the inflammatory response is both the result of a variable balance between epithelial basement membrane autoreactive T-cell-mediated inflammation, in the lesional tissues, and also results from circulating autoantibodies to basement membrane proteins. Although the effects of inflammation and scarring are closely related it is easier to dissect out the mechanisms for each separately:

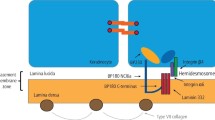
Summary of the pathogenesis of inflammation and scarring in ocular MMP. This illustrates the description in this section in the text, for which the evidence is summarised in Table 3. In brief a loss of tolerance to mucosal epithelial basement membrane proteins results in the development of pathogenic autoreactive T cells (aT) which help autoreactive B cells (aB) to proliferate in the regional lymph nodes, and differentiate into plasma cells. The latter produce circulating IgG and IgA autoantibodies to the mucosal basement membrane, which are detectable in the serum of some patients by indirect immunofluorescence (and other antibody specific assays). Plasma cells are also found in the conjunctival mucosal substantia propria where they may produce local antibody. In the mucosal epithelium direct immunofluorescence tests may show the presence of IgG and/or IgA fixed to the basement membrane where C3 (complement 3) is also identified in some patients. Activation of C3 at the basement membrane precipitates the complement cascade and acute inflammation at the basement membrane. This is the injury and inflammation phase of the disease causing an accumulation of inflammatory effector cells (neutrophils, dendritic cells, mast cells, eosinophils, macrophages and T cells) and the associated cytokines interleukin (IL) IL-2, IL-5, IL-13 and growth factors: IFNγ (interferon gamma, TNFα (tumour necrosis factor alpha), resulting in, often severe, inflammation and expansion of both T helper subset 1 and 2 cells into a chronic inflammatory response. An alternative effector pathway may be more important, in the absence of autoantibody, in some subsets of patients whereby autoreactive T cells to basement membrane components home in on the mucosa. Here they can create an inflammatory response, in the absence of antibody, through their cytokines and growth factors including IFNγ, TNFα, IL-4, IL-5 and IL-13. This inflammatory response results in fibrosis through the effects of profibrotic mediators released by macrophages, T cells, mast cells and eosinophils on fibroblasts, including PDGF (platelet derived growth factor), IL-13, TGFβ (transforming growth factor beta) and HSP47 (heat shock protein). Fibrosis also results from ALDH/RA (aldehyde dehydrogenase/retinoic acid) mediated paracrine effects of dendritic cells that activate a profibrotic phenotype in fibroblasts. During both active inflammation, and once inflammation has resolved, the activated MMP fibroblasts continue to scar as these remain profibrotic because of an ALDH/RA mediated autocrine effect. The latter probably results in RA dependent TGFβ activation and or induction, further driving fibrosis.
Inflammation
Current descriptions ascribe the inflammatory response in MMP as arising from loss of tolerance to basement membrane proteins resulting in autoreactive T cells interacting with autoreactive B cells in the regional lymph nodes. This results in the generation of plasma cells, producing IgG or IgA circulating antibodies. Antibody may also be locally produced given that plasma cells are found in the lesional tissue, whereas the evidence for the presence of B cells in lesional tissue is mixed.64, 65 Loss of tolerance is more common in individuals expressing HLA-DQB*0301, which may promote (restrict) the activation of T cells that are autoreactive to basement membrane proteins.59 Following this IgG and/or IgA antibody binds to the basement membrane in the conjunctival mucosa, resulting in complement fixation and the development of an acute inflammatory response leading to an influx of neutrophils, macrophages, dendritic cells and both cytotoxic (Tc) and helper (Th) T cells and plasma cells.64, 65 MHC class II protein (required to present peptides to the immune system) are highly expressed in MMP conjunctiva.65 Mast cells and eosinophils, capable of producing pro-fibrogenic cytokines are also found in acute disease.66 Transforming growth factor beta (TGFβ) is overexpressed in acute disease compared with controls but not in chronic disease and, although the profibrotic factors, platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) are present they are not overexpressed, suggesting that in chronic disease fibroblast activity remains functionally and morphologically abnormal after the withdrawal of the influence of growth factors.65, 67 The B7-2 costimulatory molecule required for T-cell proliferation in the presence of IL-2 is also overexpressed and can be expected to lead to increased T-cell expansion.64 Of the cytokines, IL-2, IFNγ and TNFα (Th1 signature cytokines) are all present although not overexpressed compared with controls.65, 67 However, serum TNFα, an important pro-inflammatory mediator, has been shown to be elevated in MMP compared with controls and, in a more recent study, to be overexpressed in inflamed compared with treated MMP conjunctiva.68 Evidence for the activity of Th2 cells in ocular MMP comes from the finding of elevated levels of IL-13 in inflamed MMP conjunctiva69 and IL-5 in serum,66 although the latter is also produced by mast cells.
Scarring
Profibrotic mediators have been investigated in several studies including TGFβ, PDGF, FGF and IL-13 as described above. Collagen type I and III are increased in ocular MMP stromal tissue. Cultured in vitro MMP conjunctival fibroblasts respond to TGFβ by the induction of heat shock protein 47 (HSP47), which is thought to influence procollagen synthesis and fibrosis resulting in collagen Type I production.70 Macrophage accumulation in the conjunctiva is probably an important event in the pathogenesis of MMP conjunctival scarring stimulated by macrophage derived cytokines and growth factors, including IL-4, PDGF, and TGFβ. Increased levels of macrophage inhibition factor (MIF)71 and macrophage colony stimulating factor (m-CSF),72 have been demonstrated in both in vitro cultured MMP fibroblasts and in whole conjunctiva and associated with increased numbers of macrophages. Connective tissue growth factor (CTGF), a downstream profibrotic mediator of TGF-β1, is overexpressed in both ocular MMP whole conjunctiva and in MMP cultured fibroblasts.73 These studies demonstrate that the effector cells, cytokines and growth factors necessary for fibrosis are present in the MMP conjunctiva. However, the mechanisms that relate this inflammatory milieu to the production of the extracellular matrix (ECM) by fibroblasts, that results in scarring, have not been identified.74, 75, 76 However, our recent studies outlined below have identified one control mechanism.
We have previously confirmed that OMMP fibroblasts maintain a profibrotic phenotype in vitro and have hypothesised that progressive fibrosis may be precipitated by the inflammation associated with ocular MMP, and then persist in eyes having clinical control of inflammation because of the continuing activity of persistently profibrotic fibroblasts.77 In recently published studies78 we have identified that the aldehyde dehydrogenase (ALDH)/retinoic acid (RA) metabolic pathway regulates this profibrotic activity in ocular MMP conjunctival fibroblasts in vitro. We have shown that ALDH is overexpressed in ocular MMP conjunctiva at the gene and protein level, compared with controls, and that ALDH inhibition with disulfiram abolished the profibrotic phenotype in MMP conjunctiva, resulting in the adoption of a normal control phenotype. Conversely in vitro fibroblasts from normal controls adopt a profibrotic phenotype when treated with RA, the metabolic product of ALDH. These findings provide evidence for ALDH/RA autoregulation in ocular MMP fibroblasts as a mechanism underlying progressive conjunctival scarring seen in this disease and the potential for ALDH inhibition with disulfiram as a therapy for fibrosis. These findings were further confirmed in a mouse model of ovalbumin induced severe conjunctival inflammation that was developed for allergic eye disease studies in which we have shown that conjunctival scarring develops concurrent with inflammation. ALDH inhibition in this model, using topical disulfiram, was effective in preventing scarring in vivo, and also restored in vitro in mouse conjunctival fibroblasts to a normal phenotype, as in ocular MMP. Furthermore, another paper published with this study, has shown that in the same mouse model conjunctival scarring is initiated by the key role of dendritic cells, through paracrine production of ALDH/RA effecting conjunctival fibroblasts.79 Given our hypothesis that the scarring in ocular MMP is the result of the inflammatory response in MMP, rather than due to the autoimmune pathogenesis per se, we believe this mouse model provides a surrogate for studying immune-mediated conjunctival scarring. Disulfiram is a drug already licensed for alcohol abuse control. These studies suggest that it could be repurposed for the topical treatment of conjunctival scarring in ocular MMP and provide justification for a randomized controlled trial of disulfiram therapy in this disease. Other mucosal scarring diseases are potential targets for further study using these techniques. Conjunctival diseases that may share these fibrotic mechanisms are Stevens-Johnson syndrome, atopic keratoconjunctivitis and trachoma.
Although ALDH inhibition is effective in these models, we do not understand the molecular mechanisms underlying these profibrotic effects of ALDH/RA. It is possible that these profibrotic effects are mediated by similar mechanisms to those described in liver fibrosis,8, 18 by the induction of the TGFβ1 gene, and/or activation of latent TGFβ1, for example by increased plasminogen activator levels. Active TGFβ drives critical changes in fibroblast metabolism, activation and ECM production in concert with pro-inflammatory cytokines and growth factors that affect fibroblast activity.17 Alternatively, ALDH/RA might cause its effects through altered cellular energy metabolism via activation of TGFβ or as a consequence of the modulation of the transcription of metabolic genes.33 The ALDH/RA metabolic pathway and these potential molecular mechanisms are described in Figure 10.
Treatment summary
Treatment outcomes for ocular MMP before 1980
These were poor until the 1980’s when immunomodulation techniques were introduced for their management for the first time. In the first series of cases reported by Morris in 1889 the outcomes were summarised: 28 cases (12F, 13M) onset from infancy to 76 years. ‘Disease began in the skin in 16, other mucosae in 4, and in the eye in 8. Twelve cases had conjunctival blistering, and others a pseudomembrane, entropion, progressive conjunctival shrinkage, cloudy cornea, thickened bulbar conjunctiva and xerophthalmia. Generally, vision is lost apart from perception of light although corneal perforation and destruction of the globe has been reported. One case had been described with spontaneous remission. Entropion surgery and application of lotions for the inflammation are palliative, there is no treatment for the progressive scarring. The pathology is obscure. It is a very rare disease’.80 In Swanzy’s 1895 textbook he stated ‘Treatment is helpless in respect of arresting the progress of disease, or of restoring sight when lost in consequence of it. The most one can do is to relieve the distressing symptoms by emollients to the conjunctiva, and by the use of closely fitting goggles, to protect from wind, dust and sun. Internally arsenic is indicated.81’ By 1951 Sorsby, in Systemic Ophthalmology stated of ocular pemphigus ‘Treatment is uniformly unsuccessful’.82 The first textbook I owned when I started ophthalmology in 1978 was Parsons’ Diseases of the Eye 16th Edition in which it was stated that in the treatment of Benign Mucous Membrane Pemphigoid ‘Local treatment is unavailing as also, indeed, is general treatment’.83 The use of dapsone for the management of pemphigus and pemphigoid was introduced in the 1970’s, but not for ocular MMP until 1982.84 Systemic steroids for the control of acute disease in ocular MMP were described by Mondino in 1979.85 The first use of immunosuppressive therapy, with azathioprine and cyclophosphamide, was described by Foster for two cases in 1980,86 followed by a larger series using these drugs in 1982.87 Subsequent publications have proliferated and are described below.
Current treatment
Successful management of disease demands the integration of the following:
-
1)
Control of surface disease
-
i
Blepharitis
-
ii
Dry eye
-
iii
Corneal punctate epitheliopathy
-
iv
Keratinisation
-
v
Trichiasis, entropion and lagophthalmos
-
vi
Persistent epithelial defects
-
vii
Corneal perforation
-
viii
Iatrogenic toxicity
-
i
-
2)
Control of immune mediated inflammation with systemic immunomodulation
-
3)
Control of fibrosis
-
4)
Prophylaxis of corneal ulceration and exposure
-
5)
Improving vision in patients with corneal blindness.
The ocular surface disease management, and a synopsis of management with systemic immunomodulatory therapy, and visual rehabilitation, has been discussed in detail in two reviews from our group43, 44 and a further recent review has summarized the use of immunomodulatory therapy in detail.36 In this publication I have restricted the description of treatments to a summary of the role of immunomodulatory techniques and the evidence for their use in ocular MMP, with guidelines for their use.
Use of systemic immunomodulation to control immune-mediated inflammation
Lack of effect of topical immunosuppressive therapy in ocular MMP
Topical steroid treatment is ineffective in controlling progressive ocular MMP, offering only variable symptomatic relief.38, 85 Its adverse effects of cataract and glaucoma generally outweigh the benefits. Subconjunctival steroids may be temporarily effective, but relapses occur when the injections are stopped85 and prolonged use also leads to cataract and glaucoma. Anecdotally, topical steroid will relieve discomfort and inflammation in some patients with mild/moderate disease activity but has not reduced the activity of severe inflammatory disease or the progression of scarring. Topical ciclosporin has been used in only four reported cases of whom two had some response;88 we have little experience with this for ocular MMP and it is probable that the poor results reported for systemic therapy with ciclosporin may have inhibited further investigation of this modality. As a result, systemic immunomodulatory therapy is currently the standard of care for these MMP patients.23, 89, 90
Evidence for the effect of systemic immunosuppression on progression of disease
The primary goals of treatment of ocular MMP are to control inflammation and arrest fibrosis, in order to prevent progression of disease to more advanced stages and blindness. Most cicatrisation is occurs during active inflammation,85 but despite control of inflammation in 70–78% of patients with systemic immunosuppression, progressive fibrosis was still observed in 61/115 (53%)9 and 23/54 (42%)12 of patients. Without treatment, conjunctival scarring in ocular MMP progresses in 13/20 (64%) of patients over 10–53 months.91 Progression is more frequent in the advanced stages of disease.92 Use of immunosuppressive therapy has been shown to slow progression of disease in one case series92 and control of inflammation has been shown to prevent progression in one RCT.23 With current immunosuppressive regimens, progression of cicatrisation has still been observed in 10–53% of ocular MMP patients92, 93, 94 and is more rapid in patients <60 years of age.95 A subset of MMP patients with ocular involvement have ongoing conjunctival fibrosis without overt clinical signs of inflammation.96 However, despite the absence of clinical signs of inflammation, there may still be significant cellular infiltrate on histological evaluation (‘white inflammation’).67, 97, 98 Further systemic immunosuppression with potential systemic toxicity may not necessarily be helpful in these cases, for whom more specific local therapy targeting the cellular infiltrate or fibrogenic process would be ideal. There is evidence for both ongoing residual subclinical inflammation68, 69 and transformed profibrotic fibroblasts77 as the putative drivers of scarring which progresses, despite apparent clinical control of inflammation with systemic immunosuppression. No current medical therapy is able to reverse the cicatrisation or ocular surface problems once they have developed.
Criteria for initiating immunosuppressive therapy
About 25% of OcMMP patients do not require immunosuppression93 as they have few symptoms, limited scarring, mild or no inflammation and slow progression, or are in remission. In end-stage ‘burned out’ disease, immunosuppression is also unnecessary as eyes are usually comfortable, albeit blind, and treatment only slows scarring without significantly reversing it. In these patient groups, if conjunctival incision surgery is planned, such as that required for cataract extraction when the fornices are very short, or before fornix reconstruction, then immunosuppression should be started beforehand, to prevent an exacerbation of postoperative inflammation and scarring which may be severe, and will result in a poor surgical result and disease progression. For cataract surgery through a clear corneal incision, without conjunctival surgery99 or fornix incision for subtenon’s anaesthesia, and for lid surgery without a conjunctival incision, it is not necessary in my experience, to introduce preoperative immunosuppressive therapy.
Identifying inflammation due to underlying disease rather than to the secondary effects of a poor ocular surface
Identification of inflammation that is primarily due to the disease process in MMP, rather than to inflammation secondary to the effects of ocular surface disease or topical drug therapy, is essential before initiating immunomodulatory therapy. This is rarely a problem in those patients presenting with more severe disease. However, it can be difficult in those with less severe inflammation. It is necessary to differentiate inflammation due to the associated surface disease (dry eye, blepharoconjunctivitis, and lash abrasion), from immune mediated inflammation. Clinically this can be done by evaluating the degree of bulbar conjunctival inflammation under the upper lid, which is free of the worst effects of surface disease. However, sectoral inflammation, although not common, does occur and may affect any quadrant of the bulbar conjunctiva as shown in Figure 11. Topical drop toxicity/allergy, typically resulting from glaucoma medications or preservatives may cause diffuse inflammation and be impossible to distinguish from that caused by MMP unless the topical therapy is discontinued: it takes 1-2 weeks for this to start to improve after withdrawing glaucoma medication, and any other topical drops, apart from non-preserved saline (see When to investigate? section).

Sectoral conjunctival inflammation in ocular mucous membrane pemphigoid (MMP). The same eye of a patient showing the superior bulbar conjunctiva free of inflammation but a localised area of inflammation in the inferior bulbar conjunctiva.
Evidence for the efficacy of different immunosuppressive regimens in controlling inflammation and guidelines for its delivery
Evidence for the effect of current immunosuppressive therapy in ocular MMP is summarised in Table 4 and comes from cohort studies,100, 101 interventional and retrospective case series43, 84, 92, 102, 103, 104, 105, 106, 107, 108, 109, 110 and two randomized trials.23 These have indicated a role for dapsone, sulfasalazine or sulphapyridine for mild to moderate inflammation, azathioprine, mycophenolate or methotrexate for moderate inflammation, or for disease not responding to sulphonamide therapy, and cyclophosphamide with a short course of prednisolone, for severe inflammation. Combinations of sulfas and myelosuppressive drugs (methotrexate, azathioprine, mycophenolate, and cyclophosphamide) with or without prednisolone can be effective. For patients having severe disease, unresponsive to these conventional immunosuppressants, both Rituximab, and intravenous immunoglobulin (IViG) are used. Notes on the use of these drugs, as 'step up and step down therapy', including combination therapy as favoured by UK specialists, are in Table 4 and Figure 12. Supplementary Appendix 5 includes a protocol for the administration of IVIg and Supplementary Appendix 4 is a synopsis of immunosuppression management guidelines for use in clinic.

Immunosuppression for ocular mucous membrane pemphigoid (MMP). This illustrates ‘step up’ and ‘step down’ therapy to control conjunctival inflammation as described in the section on ‘Evidence for the efficacy of different immunosuppressive regimens in controlling inflammation and guidelines for its delivery’ and in Table 4 Row 34, and Saw VP 2008.9 (a) Drugs in different coloured boxes can be combined. The drugs in the dark red boxes (cyclophosphamide, mycophenolate, azathioprine and methotrexate) are all myelosuppressives: combining these results in unacceptable toxicity. Some authors have described the use of CD 20 inhibitors (such as Rituximab) with existing myelosuppressive therapy for unresponsive ocular MMP119 but this may be unsafe and we follow the recommendations of restricting adjunctive drugs to sulfas120 and stop the myelosuppressives immediately before the first CD20 monoclonal infusion. Prednisolone can generally be given for a short term effect with any of the other drugs listed but is usually needed in high doses, with its associated side effects. (b) In general, all of these drugs, except high dose steroids take 2-4 months to take effect. This period is probably shorter for the CD 20 monoclonals than the other drugs. A full effect will often take longer and for this reason, unless inflammation is deteriorating, I do not usually alter therapy for 12 weeks. (c) For mild/moderate ocular MMP use step up therapy. Start with a sulfa and, there is no effect, or toxicity, introduce mycophenolate. If the effect is limited but the drug is tolerated, add mycophenolate to the sulfa. Azathioprine (in grey typeface) is the second line drug when mycophenolate is not tolerated and methotrexate (in grey typeface) the third line drug in this situation because these are generally less well tolerated than mycophenolate and no more effective. If none of these drugs works switch to cyclophosphamide without using adjunctive steroid unless the patient is deteriorating. Failing these then a CD20 monoclonal is the next choice of drug and, if there is a lack of response to that 2 months after a second cycle of therapy, then start IV immunoglobulin therapy. (d) For severe MMP (severe conjunctival inflammation±limbitis±corneal ulceration) use step down therapy. Start oral Prednisolone 1 mg/kg WITH cyclophosphamide 1.0–1.5 mg/kg. Very severe cases will get a short term benefit from intravenous methylprednisolone 1 g IV daily for 3 days before starting oral prednisolone with cyclophosphamide (unpublished data). If cyclophosphamide and prednisolone are not effective within 2–3 months, add a sulfa and then switch to a CD20 monoclonal. If this fails, as in the previous example, then start IVIG. Once control of inflammation has been established the patient can start stepdown therapy. (i) For cyclophosphamide follow the toxicity guidelines in Table 4 row 19, I usually maintain therapy for up to 12 months in the hope that a longer period of therapy might induce a remission. Two months before discontinuing cyclophosphamide, introduce a sulfa, if tolerated. After discontinuing cyclophosphamide patients with full control of inflammation can discontinue sulfas after another 2–4 months therapy. For patients with partial control the sulfa is maintained and mycophenolate (azathioprine or methotrexate) added. Drugs can be discontinued as required dependent on disease control and the development of side effects. ii. After CD 20 monoclonals patients may be in remission and any sulfa therapy can be discontinued after 2–4 months therapy. In the event of a relapse a decision has to be made whether to control this with step up therapy or a further cycle of a CD 20 monoclonal. After IVIG patients will usually still be in myelosuppressive agents which can be stepped down or stepped up as described above.
Remission and when to stop therapy
Once complete control of inflammation has been achieved we continue immunomodulation, if well tolerated, for at least 12 months. Following this, the dose is reduced and can be stopped, if the patient wishes, providing they understand that it will need to be recommenced if disease activity recurs. Life-long follow-up is necessary, because disease recurs in up to 1/3 of patients.105 Our series of 115 patients showed remission without therapy of at least 6 months in 20 (17%) of patients with a relapse in 4/20 (20%). In our experience long term remissions are uncommon using conventional drug therapies. However, Thorne et al using high dose cyclophosphamide and prednisolone, achieved better remission rates, of at least 3 months without therapy, in 40/44 (91%) within 2 years of initiating therapy, followed by an 8/40 (20%) relapse rate. However, we think the risks of adverse effects outweigh the benefits as these results require cumulative cyclophosphamide doses which probably exceed safe levels for the development of malignancy (see Table 4, rows 14 and 19).
The last 30 years and horizon scanning for the next decade
In the last 30 years there have been great advances in the understanding of the pathogenesis and therapy of three of the commonest causes of conjunctival scarring: trachoma,111 mucous membrane pemphigoid44 and atopic keratoconjunctivitis.5 These include the introduction of mass azithromycin delivery for trachoma control and the understanding that the scarring response in trachoma is only indirectly related to the precipitating infection resulting from the secondary profibrotic effects of infection. Also the unravelling of the pathogenesis of atopic keratoconjunctivitis and the randomised controlled trials, demonstrating the effectiveness of topical ciclosporin in its management. For ocular mucous membrane pemphigoid, the introduction of effective therapy with systemic myelosuppressives, currently the standard of care, had only just been introduced 30 years ago before which a majority of patients progressed to bilateral blindness. There was minimal understanding of the pathogenesis of MMP at that time and what we now know is feeding into the application of new therapies for inflammation. The latter include the use of monoclonal CD20 mediated B cell elimination, and the potential development of anti-scarring therapies. There are four areas in particular where advances can be expected to contribute substantially to improved outcomes for patients: improved diagnostic tests, the identification of biomarkers of disease activity, the further development of biological drugs for therapy, and the development of topical therapies for both inflammation and scarring.
Better diagnostics
The limitations of the current reliance on the detection of autoantibodies for disease identification, particularly for ocular MMP, but also for MMP affecting other sites, have been outlined above. We have some preliminary data using short peptides of BP180 that appear to be more sensitive than currently available substrates for the identification of autoantibodies in ocular MMP. An alternative approach has been used for the investigation of trachoma phenotypes using genetic biomarkers, that are up and down regulated during the inflammation, scarring and resolution.112, 113 A similar approach in MMP, and other causes of CC, may be expected to provide an alternative route to autoantibody detection, for both diagnosis and for the assessment of disease activity.
Biomarkers of disease activity
More objective biomarkers than clinical slit-lamp examination are needed to identify both inflammatory and scarring activity, to help determine the effects of therapy and guide its initiation and withdrawal. Some progress has been made using neutrophils and their products for this.97, 114 Expression of genes that are up- and downregulated in inflammation and scarring may also be worth investigating for this purpose.
Biological drugs for MMP
Conventional non biological immunomodulatory therapy with cyclophosphamide is effective anti-inflammatory therapy for between 80% to 92% of cases in the larger series. However, the CD20 monoclonal Rituximab has been increasingly used as rescue therapy for patients with severe ocular MMP who have failed cyclophosphamide therapy, with combined success rates of 80% in this group. Although efforts have been made to carry out a randomised trial of this drug in ocular MMP the cost of therapy has prohibited this, and manufacturers have been unwilling to support research into an orphan indication. However, there are numerous CD20 monoclonal biosimilars in development and it is probable that these drugs will become available for prospective case series, and randomised controlled trials. The latter are needed to establish the effect of this drug in a number of areas. These include the effect in early ocular MMP, as opposed to unresponsive later stage disease. The length of remission, evidence for the induction of tolerance to autoantigens, and the identification of responders and non-responders needs to be investigated, as has been done in pemphigus.115 The optimum treatment regimen needs to be established, and the side effect profile compared with that of the other drugs used in this, relatively elderly, patient group having MMP.
Topical therapies for inflammation and scarring
There is an unmet need for effective topical therapies for inflammation and scarring to eliminate the morbidity associated with the side effects of systemic therapies. There are potential new topical therapies for both scarring and inflammation in CC. We have outlined the evidence for a promising new therapy, using ALDH inhibition, which has the potential to control scarring with topical therapy. ALDH inhibition is also in development elsewhere as an anti-inflammatory therapy for allergic eye disease and anterior uveitis. Rapidly effective anti-scarring therapy for CC is critical in preventing morbidity, given the speed of scarring during severe inflammatory episodes which take 2–4 months to resolve with current anti-inflammatory therapies in ocular MMP, and which are also very rapid in diseases like Stevens-Johnson syndrome. The latter is another additional target disease for topical anti-scarring therapy which will be effective during acute phase inflammation, when most of the scarring occurs. Perfenidone is a new class of immunosuppressant with anti-inflammatory and antifibrotic effects. Its mechanisms of action are not fully established. In different model systems it has been shown to abrogate TGF-β1-stimulated collagen synthesis by inhibiting the upregulation of HSP47 and Col1 RNA, it blocks the proliferative effects of PDGF; reduces fibroblast proliferation and downregulates the proinflammatory cytokines, TNFα, IFNγ, IL-1B and IL-6.116 Topical application has been studied in human scleroderma117 and in glaucoma scarring models.118
Finally, the topical use of anti-inflammatory monoclonal antibodies, such as the anti TNFα drugs that could be given subconjunctivally, should be evaluated.
Although a lot has been done in the last 30 years, the tools for understanding the pathogenesis of these diseases have expanded exponentially, and the potential for new therapies has seldom been greater, for patients cursed with these diseases.
References
Power H . Prefatory Memoir. In Burdon-Sanderson J, Hulke JW (eds The Collected Papers of Sir William Bowman, Bart., F.R.S.. Vol. 2 Harrison and Sons: London, 1892. pp xi–xxiii.
James RR . British masters of ophthalmology series: 16.-Sir William Bowman, Bart., F.R.S., 1816-1892. Br J Ophthalmol 1925; 9: 481–494.
Thorne JE, Anhalt GJ, Jabs DA . Mucous membrane pemphigoid and pseudopemphigoid. Ophthalmology 2004; 111: 45–52.
Radford CF, Rauz S, Williams GP, Saw VP, Dart JK . Incidence, presenting features, and diagnosis of cicatrising conjunctivitis in the United Kingdom. Eye (Lond) 2012; 26: 1199–1208.
Guglielmetti S, Dart JK, Calder V . Atopic keratoconjunctivitis and atopic dermatitis. Curr Opin Allergy Clin Immunol 2010; 10: 478–485.
Shikari H, Antin JH, Dana R . Ocular graft-versus-host disease: a review. Surv Ophthalmol 2013; 58: 233–251.
O'Donnell JJ Jr., Karakus S, Doroslovacki P, Akpek EK . Sarcoidosis Presenting with Cicatrizing Conjunctivitis. Optom Vis Sci 2015; 92: e173–e175.
Saw VP, Dart JK, Sitaru C, Zillikens D . Cicatrising conjunctivitis with anti-basement membrane autoantibodies in ectodermal dysplasia. Br J Ophthalmol 2008; 92: 1403–1410.
Saw VP, Dart JK, Rauz S, Ramsay A, Bunce C, Xing W et al. Immunosuppressive therapy for ocular mucous membrane pemphigoid strategies and outcomes. Ophthalmology 2008; 115: 253–261.
Sullivan BD, Crews LA, Sonmez B, de la Paz MF, Comert E, Charoenrook V et al. Clinical utility of objective tests for dry eye disease: variability over time and implications for clinical trials and disease management. Cornea 2012; 31: 1000–1008.
Alvarenga LS, Mannis MJ . Ocular Rosacea. Ocul Surf 2005; 3: 41–58.
Williams GP, Radford C, Nightingale P, Dart JK, Rauz S . Evaluation of early and late presentation of patients with ocular mucous membrane pemphigoid to two major tertiary referral hospitals in the United Kingdom. Eye (Lond) 2011; 25: 1207–1218.
Ahuero AE, Jakobiec FA, Bhat P, Ciralsky JB, Papaliodis GN . Paraneoplastic conjunctival cicatrization: two different pathogenic types. Ophthalmology 2010; 117: 659–664.
Chirinos-Saldana P, Zuniga-Gonzalez I, Hernandez-Camarena JC, Navas A, Ramirez-Luquin T, Robles-Contreras A et al. Cicatricial changes in ocular pemphigus. Eye (Lond) 2014; 28: 459–465.
Spaide RF, Ward DL . Conjunctival biopsy in the diagnosis of sarcoidosis. Br J Ophthalmol 1990; 74: 469–471.
Servat JJ, Bernardino CR . Effects of common topical antiglaucoma medications on the ocular surface, eyelids and periorbital tissue. Drugs Aging 2011; 28: 267–282.
Dart J . Corneal toxicity: the epithelium and stroma in iatrogenic and factitious disease. Eye (Lond) 2003; 17: 886–892.
Schmidt E, Zillikens D . Pemphigoid diseases. Lancet 2013; 381: 320–332.
Pohla-Gubo G, Hintner H . Direct and indirect immunofluorescence for the diagnosis of bullous autoimmune diseases. Dermatol Clin 2011; 29: 365–372 vii.
Baum S, Sakka N, Artsi O, Trau H, Barzilai A . Diagnosis and classification of autoimmune blistering diseases. Autoimmun Rev 2014; 13: 482–489.
Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol 2002; 138: 370–379.
Nayar M, Wojnarowska F, Venning V, Taylor CJ . Association of autoimmunity and cicatricial pemphigoid: is there an immunogenetic basis? J Am Acad Dermatol 1991; 25: 1011–1015.
Foster CS . Cicatricial pemphigoid. Trans Am Ophthalmol Soc 1986; 84: 527–663.
Bernauer W, Elder MJ, Leonard JN, Wright P, Dart JK . The value of biopsies in the evaluation of chronic progressive conjunctival cicatrisation. Graefes Arch Clin Exp Ophthalmol 1994; 232: 533–537.
Power WJ, Neves RA, Rodriguez A, Dutt JE, Foster CS . Increasing the diagnostic yield of conjunctival biopsy in patients with suspected ocular cicatricial pemphigoid. Ophthalmology 1995; 102: 1158–1163.
Grau AE, Setterfield J, Saw VP . How to do conjunctival and buccal biopsies to investigate cicatrising conjunctivitis: improving the diagnosis of ocular mucous membrane pemphigoid. Br J Ophthalmol 2013; 97: 530–531.
Power WJ, Tugal-Tutkun I, Foster CS . Long-term follow-up of patients with atopic keratoconjunctivitis. Ophthalmology 1998; 105: 637–642.
Magro CM, Roberts-Barnes J, Crowson AN . Direct immunofluorescence testing in the diagnosis of immunobullous disease, collagen vascular disease, and vascular injury syndromes. Dermatol Clin 2012; 30: 763–798 viii.
Lever WF . Pemphigus. Medicine 1953; 32: 1–123.
Thorne JE, Jabs DA, Nikolskaia O, Anhalt G, Nousari HC . Discoid lupus erythematosus and cicatrizing conjunctivitis: clinicopathologic study of two cases. Ocul Immunol Inflamm 2009; 10: 287–292.
Park AJ, Webster GF, Penne RB, Raber IM . Porphyria cutanea tarda presenting as cicatricial conjunctivitis. Am J Ophthalmol 2002; 134: 619–621.
Leonard JN, Kaufman D, McNab A, McKelvie P . Immunofluorescent studies in ocular cicatricial pemphigoid. Br J Dermatol 1988; 118: 209–217.
De Rojas MV, Dart JK, Saw VP . The natural history of Stevens Johnson syndrome: patterns of chronic ocular disease and the role of systemic immunosuppressive therapy. Br J Ophthalmol 2007; 91: 1048–1053.
Butt Z, Kaufman D, McNab A, McKelvie P . Drug-induced ocular cicatricial pemphigoid: a series of clinico-pathological reports. Eye 1998; 12 (Pt 2): 285–290.
Tauber J . Ocular cicatricial pemphigoid. Ophthalmology 2008; 115: 1639–1640 author reply 1640–1631.
Dart JK, Saw VP Immune modulation in ocular mucous membrane pemphigoid In Pleyer U, Alió J, Barisani- Asenbauer T, Le Hoang P, Rao NA eds Immune Modulation and Anti-Inflammatory Therapy in Ocular Disorders IOIS Guidelines. Springer: Berlin, Heidelberg, 2014. pp 19–37.
Leenutaphong V, von Kries R, Plewig G . Localized cicatricial pemphigoid (Brunsting-Perry): electron microscopic study. J Am Acad Dermatol 1989; 21: 1089–1093.
Hardy KM, Perry HO, Pingree GC, Kirby TJ Jr . Benign mucous membrane pemphigoid. Arch Dermatol 1971; 104: 467–475.
De D, Khullar G, Handa S, Saikia UN, Radotra BD, Saikia B et al. Clinical, demographic and immunopathological spectrum of subepidermal autoimmune bullous diseases at a tertiary center: a 1-year audit. Indian J Dermatol Venereol Leprol 2016; 82: 358.
Jin P, Shao C, Ye G . Chronic bullous dermatoses in China. Int J Dermatol 1993; 32: 89–92.
Wong SN, Chua SH . Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol 2002; 147: 476–480.
Elder MJ, Bernauer W, Leonard J, Dart JK . Progression of disease in ocular cicatricial pemphigoid. Br J Ophthalmol 1996; 80: 292–296.
Saw VPJ, Dart JKG, Reinhard TLF Management of ocular mucous membrane pemphigoid. In: Kriegelstein GK, Weinreb RN eds Cornea and External Eye Disease. Springer: Berlin, Heidelberg, 2008. pp 154–175.
Saw VP, Dart JK . Ocular mucous membrane pemphigoid: diagnosis and management strategies. Ocul Surf 2008; 6: 128–142.
Setterfield J, Theron J, Vaughan RW, Welsh KI, Mallon E, Wojnarowska F et al. Mucous membrane pemphigoid: HLA-DQB1*0301 is associated with all clinical sites of involvement and may be linked to antibasement membrane IgG production. Br J Dermatol 2001; 145: 406–414.
Chan LS, Soong HK, Foster CS, Hammerberg C, Cooper KD . Ocular cicatricial pemphigoid occurring as a sequela of Stevens-Johnson syndrome. JAMA 1991; 266: 1543–1546.
Tauber J, Melamed S, Foster CS . Glaucoma in patients with ocular cicatricial pemphigoid. Ophthalmology 1989; 96: 33–37.
Black AP, Seneviratne SL, Jones L, King AS, Winsey S, Arsecularatne G et al. Rapid effector function of circulating NC16A-specific T cells in individuals with mucous membrane pemphigoid. Br J Dermatol 2004; 151: 1160–1164.
Setterfield J, Shirlaw PJ, Bhogal BS, Tilling K, Challacombe SJ, Black MM . Cicatricial pemphigoid: serial titres of circulating IgG and IgA antibasement membrane antibodies correlate with disease activity. Br J Dermatol 1999; 140: 645–650.
Hertl M . Humoral and cellular autoimmunity in autoimmune bullous skin disorders. Int Arch Allergy Immunol 2000; 122: 91–100.
Murphy K, Weaver C . Janeway's Immunobiology. Garland Science, New York and Oxford 2016. pp 652–669.
Torchia D, Caproni M, Volpi W, Fabbri P . Naturally occurring regulatory T cells in mucous membrane pemphigoid lesions. Acta Dermatovenerol Alp Pannonica Adriat 2009; 18: 3–6.
Lutz HU, Miescher S . Natural antibodies in health and disease: an overview of the first international workshop on natural antibodies in health and disease. Autoimmun Rev 2008; 7: 405–409.
Prussmann W, Prussmann J, Koga H, Recke A, Iwata H, Juhl D et al. Prevalence of pemphigus and pemphigoid autoantibodies in the general population. Orphan J Rare Dis 2015; 10: 63.
Wieland CN, Comfere NI, Gibson LE, Weaver AL, Krause PK, Murray JA . Anti-bullous pemphigoid 180 and 230 antibodies in a sample of unaffected subjects. Arch Dermatol 2010; 146: 21–25.
van Beek N, Rentzsch K, Probst C, Komorowski L, Kasperkiewicz M, Fechner K et al. Serological diagnosis of autoimmune bullous skin diseases: prospective comparison of the BIOCHIP mosaic-based indirect immunofluorescence technique with the conventional multi-step single test strategy. Orphan J Rare Dis 2012; 7: 49.
Desai N, Allen J, Ali I, Venning V, Wojnarowska F . Autoantibodies to basement membrane proteins BP180 and BP230 are commonly detected in normal subjects by immunoblotting. Australas J Dermatol 2008; 49: 137–141.
Hachisuka H, Kurose K, Karashima T, Mori O, Maeyama Y . Serum from normal elderly individuals contains anti-basement membrane zone antibodies. Arch Dermatol 1996; 132: 1201–1205.
Budinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest 1998; 102: 2082–2089.
Thorlacius-Ussing G, Sorensen JF, Wandall HH, Pedersen AE . Auto-reactive T cells revised. Overestimation based on methodology? J Immunol Methods 2015; 420: 56–59.
Rosser EC, Mauri C . Regulatory B cells: origin, phenotype, and function. Immunity 2015; 42: 607–612.
Kabuto M, Fujimoto N, Takahashi T, Tanaka T . Decreased level of IL-10-producing B cells in patients with pemphigus but not with pemphigoid. Br J Dermatol 2016. e-pub ahead of print 7 October 2016 doi:10-111/bjd.15113.
Marino E, Grey ST . B cells as effectors and regulators of autoimmunity. Autoimmunity 2012; 45: 377–387.
Tesavibul N, Dorfman D, Sangwan VS, Christen W, Panayotis Z, Rojas B et al. Costimulatory molecules in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci 1998; 39: 982–988.
Bernauer W, Wright P, Dart JK, Leonard JN, Lightman S . The conjunctiva in acute and chronic mucous membrane pemphigoid. Ophthalmology 1993; 100: 339–346.
Letko E, Bhol K, Colon J, Foster CS, Ahmed AR . Biology of interleukin-5 in ocular cicatricial pemphigoid. Graefes Arch Clin Exp Ophthalmol 2002; 240: 565–569.
Elder MJ, Dart JK, Lightman S . Conjunctival fibrosis in ocular cicatricial pemphigoid—the role of cytokines. Exp Eye Res 1997; 65: 165–176.
Saw VP, Dart RJ, Galatowicz G, Daniels JT, Dart JK, Calder VL . Tumor necrosis factor-alpha in ocular mucous membrane pemphigoid and its effect on conjunctival fibroblasts. Invest Ophthalmol Vis Sci 2009; 50: 5310–5317.
Saw VP, Offiah I, Dart RJ, Galatowicz G, Dart JK, Daniels JT et al. Conjunctival interleukin-13 expression in mucous membrane pemphigoid and functional effects of interleukin-13 on conjunctival fibroblasts in vitro. Am J Pathol 2009; 175: 2406–2415.
Razzaque MS, Foster CS, Ahmed AR . Role of collagen-binding heat shock protein 47 and transforming growth factor-beta1 in conjunctival scarring in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci 2003; 44: 1616–1621.
Razzaque MS, Foster CS, Ahmed AR . Role of macrophage migration inhibitory factor in conjunctival pathology in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci 2004; 45: 1174.
Razzaque MS, Foster CS, Ahmed AR . Role of enhanced expression of m-CSF in conjunctiva affected by cicatricial pemphigoid. Invest Ophthalmol Vis Sci 2002; 43: 2977–2983.
Razzaque MS, Foster CS, Ahmed AR . Role of connective tissue growth factor in the pathogenesis of conjunctival scarring in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci 2003; 44: 1998.
Kasperkiewicz M, Zillikens D, Schmidt E . Pemphigoid diseases: pathogenesis, diagnosis, and treatment. Autoimmunity 2012; 45: 55–70.
Kourosh AS, Yancey KB . Pathogenesis of mucous membrane pemphigoid. Dermatol Clin 2011; 29: 479–484 x.
Kirzhner M, Jakobiec FA . Ocular cicatricial pemphigoid: a review of clinical features, immunopathology, differential diagnosis, and current management. Semin Ophthalmol 2011; 26: 270–277.
Saw VP, Schmidt E, Offiah I, Galatowicz G, Zillikens D, Dart JK et al. Profibrotic phenotype of conjunctival fibroblasts from mucous membrane pemphigoid. Am J Pathol 2011; 178: 187–197.
Ahadome SD, Abraham DJ, Rayapureddi S, Saw VP, Saban DR, Calder VL et al. Aldehyde dehydrogenase inhibition blocks mucosal fibrosis in human and mouse ocular scarring. JCI Insight 2016; 1: e87001.
Ahadome SD, Mathew R, Reyes NJ, Mettu PS, Cousins SW, Calder VL et al. Classical dendritic cells mediate fibrosis directly via the retinoic acid pathway in severe eye allergy. JCI Insight 2016; 1: e87012.
Morris MRH . Pemphigus of the skin and mucous membranes of the mouth associated with "essentil shrinking" and pemphigus of the conjunctivae". Br J Dermatol 1889; 1: 175–181.
Swanzy HR . A Handbook of the Diseases of the Eye and their Treatment. HK Lewis: London, 1895.
Sorsby A . Systemic Ophthalmology. Butterworth and Mosby: London and St Louis, 1951.
Miller S . Parson's Diseases Of The Eye. Churchill LIvingstone: Edinburgh, London, New York, NY, USA, 1978.
Rogers RS III, Seehafer JR, Perry HO . Treatment of cicatricial (benign mucous membrane) pemphigoid with dapsone. J Am Acad Dermatol 1982; 6: 215–223.
Mondino BJ, Brown SI, Lempert S, Jenkins MS . The acute manifestations of ocular cicatricial pemphigoid: diagnosis and treatment. Ophthalmology 1979; 86: 543–555.
Foster CS . Immunosuppressive Therapy for External Ocular Inflammatory Disease. Ophthalmology 1980; 87: 140–150.
Stephen Foster C, Wilson LA, Ekins MB . Immunosuppressive therapy for progressive ocular cicatncial pemphigoid. Ophthalmology 1982; 89: 340–353.
Holland EJ, Olsen TW, Ketcham JM, Florine C, Krachmer JH, Purcell JJ et al. Topical cyclosporin A in the treatment of anterior segment inflammatory disease. Cornea 1993; 12: 413–419.
Sacher C, Hunzelmann N . Cicatricial pemphigoid (mucous membrane pemphigoid): current and emerging therapeutic approaches. Am.J Clin.Dermatol. 2005; 6: 93–103.
Chang JH, McCluskey PJ . Ocular cicatricial pemphigoid: manifestations and management. Curr Allergy Asthma Rep 2005; 5: 333–338.
Mondino BJ, Brown SI . Ocular cicatricial pemphigoid. Ophthalmology 1981; 88: 95–100.
Mondino BJ, Brown SI . Immunosuppressive therapy in ocular cicatricial pemphigoid. Am J Ophthalmol 1983; 96: 453–459.
Elder MJ, Bernauer W, Leonard J, Dart JK . Progression of disease in ocular cicatricial pemphigoid. Br J Ophthalmol 1996; 80: 292–296.
Miserocchi E, Baltatzis S, Roque MR, Ahmed AR, Foster CS . The effect of treatment and its related side effects in patients with severe ocular cicatricial pemphigoid. Ophthalmology 2002; 109: 111–118.
Rauz S, Maddison PG, Dart JK . Evaluation of mucous membrane pemphigoid with ocular involvement in young patients. Ophthalmology 2005; 112: 1268–1274.
Elder MJ . The role of cytokines in chronic progressive conjunctival cicatrisation. Dev Ophthalmol 1997; 28: 159–175.
Williams GP, Nightingale P, Southworth S, Denniston AK, Tomlins PJ, Turner S et al. Conjunctival neutrophils predict progressive scarring in ocular mucous membrane pemphigoid. Invest Ophthalmol Vis Sci 2016; 57: 5457–5469.
Bernauer W, Wright P, Dart JK, Leonard JN, Lightman S . The conjunctiva in acute and chronic mucous membrane pemphigoid. An immunohistochemical analysis. Ophthalmology 1993; 100: 339–346.
Geerling G, Dart JK . Management and outcome of cataract surgery in ocular cicatricial pemphigoid. Graefes Arch Clin Exp Ophthalmol 2000; 238: 112–118.
Foster CS, Wilson LA, Ekins MB . Immunosuppressive therapy for progressive ocular cicatricial pemphigoid. Ophthalmology 1982; 89: 340–353.
Mondino BJ . Cicatricial pemphigoid and erythema multiforme. Ophthalmology 1990; 97: 939–952.
Doan S, Lerouic JF, Robin H, Prost C, Savoldelli M, Hoang-Xuan T . Treatment of ocular cicatricial pemphigoid with sulfasalazine. Ophthalmology 2001; 108: 1565–1568.
Elder MJ, Leonard J, Dart JK . Sulphapyridine—a new agent for the treatment of ocular cicatricial pemphigoid. Br J Ophthalmol 1996; 80: 549–552.
Elder MJ, Lightman S, Dart JK . Role of cyclophosphamide and high dose steroid in ocular cicatricial pemphigoid. Br J Ophthalmol 1995; 79: 264–266.
Foster CS, Neumann R, Tauber J . Long-term results of systemic chemotherapy for ocular cicatricial pemphigoid. Doc Ophthalmol 1992; 82: 223–229.
Letko E, Ahmed AR, Foster CS . Treatment of ocular cicatricial pemphigoid with tacrolimus (FK 506). Graefes Arch Clin Exp Ophthalmol 2001; 239: 441–444.
McCluskey P, Chang JH, Singh R, Wakefield D . Methotrexate therapy for ocular cicatricial pemphigoid. Ophthalmology 2004; 111: 796–801.
Neumann R, Tauber J, Foster CS . Remission and recurrence after withdrawal of therapy for ocular cicatricial pemphigoid. Ophthalmology 1991; 98: 858–862.
Tauber J, Sainz DLM, Foster CS . Systemic chemotherapy for ocular cicatricial pemphigoid. Cornea 1991; 10: 185–195.
Thorne JE, Woreta FA, Jabs DA, Anhalt GJ . Treatment of ocular mucous membrane pemphigoid with immunosuppressive drug therapy. Ophthalmology 2008; 115: 2146–2152 e2141.
Taylor HR . Doyne Lecture: trachoma, is it history? Eye (Lond) 2009; 23: 2007–2022.
Holland MJ, Jeffries D, Pattison M, Korr G, Gall A, Joof H et al. Pathway-focused arrays reveal increased matrix metalloproteinase-7 (matrilysin) transcription in trachomatous trichiasis. Invest Ophthalmol Vis Sci 2010; 51: 3893–3902.
Burton MJ, Bailey RL, Jeffries D, Mabey DC, Holland MJ . Cytokine and fibrogenic gene expression in the conjunctivas of subjects from a Gambian community where trachoma is endemic. Infect Immun 2004; 72: 7352–7356.
Arafat SN, Suelves AM, Spurr-Michaud S, Chodosh J, Foster CS, Dohlman CH et al. Neutrophil collagenase, gelatinase, and myeloperoxidase in tears of patients with stevens-johnson syndrome and ocular cicatricial pemphigoid. Ophthalmology 2014; 121: 79–87.
Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, Lim A et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med 2013; 5: 175ra130.
Selvaggio AS, Noble PW . Pirfenidone Initiates a New Era in the Treatment of Idiopathic Pulmonary Fibrosis. Annu Rev Med 2016; 67: 487–495.
Rodriguez-Castellanos M, Tlacuilo-Parra A, Sanchez-Enriquez S, Velez-Gomez E, Guevara-Gutierrez E . Pirfenidone gel in patients with localized scleroderma: a phase II study. Arthritis Res Ther 2015; 16: 510.
Zhong H, Sun G, Lin X, Wu K, Yu M . Evaluation of pirfenidone as a new postoperative antiscarring agent in experimental glaucoma surgery. Invest Ophthalmol Vis Sci 2011; 52: 3136–3142.
Doan S, Roux-Villet C, Prost-Squarcioni C, Caux F, Hoang-Xuan T . Rituximab in Severe Ocular Cicatricial Pemphigoid. ARVO Meeting Abstracts 2006; 47: 594.
Canizares MJ, Smith DI, Conners MS, Maverick KJ, Heffernan MP . Successful treatment of mucous membrane pemphigoid with etanercept in 3 patients. Arch Dermatol 2006; 142: 1457–1461.
Loeffler K, Perlman J . Diffuse intraepithelial sebaceous carcinoma of the conjunctiva. Br J Ophthalmol 1997; 81: 168.
Foster CS, Calonge M . Atopic Keratoconjunctivitis. Ophthalmology 1990; 97: 992–1000.
Taylor HR, Burton MJ, Haddad D, West S, Wright H . Trachoma. Lancet 2014; 384: 2142–2152.
Rajak SN, Habtamu E, Weiss HA, Bedri A, Gebre T, Bailey RL et al. The clinical phenotype of trachomatous trichiasis in Ethiopia: not all trichiasis is due to entropion. Invest Ophthalmol Vis Sci 2011; 52: 7974–7980.
Chintakuntlawar AV, Chodosh J . Cellular and tissue architecture of conjunctival membranes in epidemic keratoconjunctivitis. Ocul Immunol Inflamm 2010; 18: 341–345.
Hammer LH, Perry HD, Donnenfeld ED, Rahn EK . Symblepharon formation in epidemic keratoconjunctivitis. Cornea 1990; 9: 338–340.
Tullo AB, Higgins PG . An outbreak of adenovirus keratoconjunctivitis in bristol. Br J Ophthalmol 1979; 63: 621–626.
Wright P . Cicatrizing conjunctivitis. Trans Ophthalmol Soc UK 1986; 105 (Pt 1): 1–17.
Mendez CA, Singh AD . Radiation therapy: anterior segment complications. Developments in ophthalmology 2013; 52: 102–113.
Creamer D, Walsh SA, Dziewulski P, Exton LS, Lee HY, Dart JK et al. U.K. guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults 2016. Br J Dermatol 2016; 174: 1194–1227.
Mitsuya J, Hara H, Ito K, Ishii N, Hashimoto T, Terui T . Metastatic ovarian carcinoma-associated subepidermal blistering disease with autoantibodies to both the p200 dermal antigen and the gamma 2 subunit of laminin 5 showing unusual clinical features. Br J Dermatol 2008; 158: 1354–1357.
Solomon l, Helm TN, Stevens C, Neiders ME, Kumar V . Clinical and immunopathologic findings in oral lichen planus pemphigoides. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007; 103: 808–813.
Henry S . Recognizing presentations of pemphigoid gestationis: a case study. Case Rep Obstet Gynecol 2014; 2014: 415163.
Venning VA, Frith PA, Bron AJ, Millard PR, Wojnarowska F . Mucosal involvement in bullous and cicatricial pemphigoid. A clinical and immunopathological study. Br J Dermatol 1988; 118: 7–15.
Daoud YJ, Foster CS, Razzaque Ahmed A . Eyelid Skin Involvement in Pemphigus Foliaceus. Ocul Immunol Inflamm 2009; 13: 389–394.
Broussard KC, Leung TG, Moradi A, Thorne JE, Fine JD . Autoimmune bullous diseases with skin and eye involvement: cicatricial pemphigoid, pemphigus vulgaris, and pemphigus paraneoplastica. Clin Dermatol 2016; 34: 205–213.
Karamursel Akpek E, Merchant A, Pinar V, Foster CS . Ocular Rosacea. Ophthalmology 1997; 104: 1863–1867.
Gorouhi F, Davari P, Fazel N . Cutaneous and mucosal lichen planus: a comprehensive review of clinical subtypes, risk factors, diagnosis, and prognosis. Sci World J 2014; 2014: 742826.
Thorne JE, Jabs DA, Nikolskaia OV, Mimouni D, Anhalt GJ, Nousari HC . Lichen planus and cicatrizing conjunctivitis: characterization of five cases. Am J Ophthalmol 2003; 136: 239–243.
Burge SM, Frith PA, Juniper RP, Wojnarowska F . Mucosal involvement in systemic and chronic cutaneous lupus erythematosus. Br J Dermatol 1989; 121: 727–741.
Bernauer W, Wright P, Dart JK, Leonard JN, Lightman S . Cytokines in the conjunctiva of acute and chronic mucous membrane pemphigoid: an immunohistochemical analysis. Graefes Arch Clin Exp Ophthalmol 1993; 231: 563–570.
Lee SJ, Li Z, Sherman B, Foster CS . Serum levels of tumor necrosis factor-alpha and interleukin-6 in ocular cicatricial pemphigoid. Invest Ophthalmol Vis Sci 1993; 34: 3522–3525.
Reiche L, Wojnarowska F, Mallon E . Combination therapy with nicotinamide and tetracyclines for cicatricial pemphigoid: further support for its efficacy. Clin Exp Dermatol 1998; 23: 254–257.
Knight A, Askling J, Granath F, Sparen P, Ekbom A . Urinary bladder cancer in Wegener's granulomatosis: risks and relation to cyclophosphamide. Ann Rheum Dis 2004; 63: 1307–1311.
Bernatsky S, Ramsey-Goldman R, Clarke AE . Malignancies and cyclophosphamide exposure in Wegener's granulomatosis. J Rheumatol 2008; 35: 11–13.
Durrani K, Papaliodis GN, Foster CS . Pulse IV cyclophosphamide in ocular inflammatory disease: efficacy and short-term safety. Ophthalmology 2004; 111: 960–965.
Harper L, Morgan MD, Walsh M, Hoglund P, Westman K, Flossmann O et al. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Ann Rheum Dis 2012; 71: 955–960.
Zurdel J, Aboalchamat B, Zierhut M, Stubiger N, Bialasiewicz A, Engelmann K . [Early clinical results with mycophenolate mofetil in immunosuppressive therapy of ocular pemphigoid]. Klin Monatsbl Augenheilkd 2001; 218: 222–228.
Letko E, Miserocchi E, Daoud YJ, Christen W, Foster CS, Ahmed AR . A nonrandomized comparison of the clinical outcome of ocular involvement in patients with mucous membrane (cicatricial) pemphigoid between conventional immunosuppressive and intravenous immunoglobulin therapies. Clin Immunol 2004; 111: 303–310.
Sami N, Letko E, Androudi S, Daoud Y, Foster CS, Ahmed AR . Intravenous immunoglobulin therapy in patients with ocular-cicatricial pemphigoid: a long-term follow-up. Ophthalmology 2004; 111: 1380–1382.
John H, Whallett A, Quinlan M . Successful biologic treatment of ocular mucous membrane pemphigoid with anti-TNF-alpha. Eye (Lond) 2007; 21: 1434–1435.
Prey S, Robert PY, Drouet M, Sparsa A, Roux C, Bonnetblanc JM et al. Treatment of ocular cicatricial pemphigoid with the tumour necrosis factor alpha antagonist etanercept. Acta Derm Venereol 2007; 87: 74–75.
Sacher C, Rubbert A, Konig C, Scharffetter-Kochanek K, Krieg T, Hunzelmann N . Treatment of recalcitrant cicatricial pemphigoid with the tumor necrosis factor alpha antagonist etanercept. J Am Acad Dermatol 2002; 46: 113–115.
Heffernan MP, Bentley DD . Successful treatment of mucous membrane pemphigoid with infliximab. Arch Dermatol 2006; 142: 1268–1270.
Le Roux-Villet C, Prost-Squarcioni C, Alexandre M, Caux F, Pascal F, Doan S et al. Rituximab for patients with refractory mucous membrane pemphigoid. Arch Dermatol 2011; 147: 843–849.
Taverna JA, Lerner A, Bhawan J, Demierre MF . Successful adjuvant treatment of recalcitrant mucous membrane pemphigoid with anti-CD20 antibody rituximab. J Drugs Dermatol 2007; 6: 731–732.
Ross AH, Jaycock P, Cook SD, Dick AD, Tole DM . The use of rituximab in refractory mucous membrane pemphigoid with severe ocular involvement. Br J Ophthalmol 2009; 93: 421–422 548.
Schumann T, Schmidt E, Booken N, Goerdt S, Goebeler M . Successful treatment of mucous membrane pemphigoid with the anti-CD-20 antibody rituximab. Acta Derm Venereol 2009; 89: 101–102.
Lourari S, Herve C, Doffoel-Hantz V, Meyer N, Bulai-Livideanu C, Viraben R et al. Bullous and mucous membrane pemphigoid show a mixed response to rituximab: experience in seven patients. J Eur Acad Dermatol Venereol 2010; 25: 1238–1240.
Rubsam A, Stefaniak R, Worm M, Pleyer U . Rituximab preserves vision in ocular mucous membrane pemphigoid. Exp Opin Biol Ther 2015; 15: 927–933.
Maley A, Warren M, Haberman I, Swerlick R, Kharod-Dholakia B, Feldman R . Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP). J Am Acad Dermatol 2016; 74: 835–840.
Foster CS, Chang PY, Ahmed AR . Combination of rituximab and intravenous immunoglobulin for recalcitrant ocular cicatricial pemphigoid: a preliminary report. Ophthalmology 2010; 117: 861–869.
Papaliodis GN, Chu D, Foster CS . Treatment of ocular inflammatory disorders with daclizumab. Ophthalmology 2003; 110: 786–789.
Sobolewska B, Deuter C, Zierhut M . Current medical treatment of ocular mucous membrane pemphigoid. Ocul Surf 2013; 11: 259–266.
Acknowledgements
For the following illustrations: Dr Jane Setterfield for Figure 5 mucosal lichen planus, and Figure 7b, d–f. Professor Mike Gleeson for Figure 7c, Professor Enno Schmidt for pemphigus vulgaris in Figure 5. Dr John Mee, of the St John's Institute of Dermatology Immunofluorescence Laboratory, for the DIF images of pemphigus, pemphigoid and the IIF image in Figure 5. For review of the pathogenesis section of the manuscript: Professor Enno Schmidt, Professor Detlef Zillikens, Dr Virginia Calder and Dr Danny Saban. For their review of the entire manuscript Dr Saaeha Rauz and Mr Ewan Craig. This work is the result of collaboration with a lot of individuals: Dr J Setterfield and Professor M Gleeson of Guys and St Thomas’s Hospital and King’s College London, Professor V Lund and Mr G Sandhu of the UCL Royal National ENT Hospital, Ms S Rauz and Mr G Williams of the Institute of Inflammation and Ageing, University of Birmingham, Dr E Schmidt, Professor D Zillikens of the Dept of Dermatology, Luebeck, Germany, Dr Chuan-Hui Kuo and Dr K Morohoshi of the Department of Ophthalmology, Division of Allergy and Immunology, Cincinnati Children's Hospital, USA, Mr R Keeler, Archivist at the Royal College of Ophthalmologists, Professor J Daniels, Dr V Calder and Dr S Ahadome of the UCL Institute of Ophthalmology, Professor D Abraham, Dr J Norman and Mr S Rayurapeddi of the Research Department of Inflammation, Division of Medicine, UCL Royal Free, Dr W Bernauer, Mr M Elder, Dr V Saw, Dr C Bunce, Sister M. Mason, and Dr C Radford of Moorfields Eye Hospital NHS Foundation Trust, Dr D Minassian of Epivision, UK and Mrs A Dart. Also, many other members of Staff at Moorfields Eye Hospital Foundation Trust and the UCL Institute of Ophthalmology. The studies could not have been done without the wholehearted support of patients with MMP and their contributions of time and tissue for research. The studies that are reported here, from me and my collaborators, have been generously funded by the following businesses, charities and individuals: UCL Business, Fight for Sight, NIHR Biomedical Research Centre for Ophthalmology, Action Medical Research, Moorfields League of Friends, Moorfields Special Trustees (now Moorfields Eye Charities) and Anonymous Donors for this research through Moorfields Eye Charities and Guide Dogs for the Blind. Lastly to Chantal Botha and the staff at Springer Nature for their help preparing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
JK Dart is involved with the development of ALDH inhibition for scarring under the Patent PCT/GB2015/051292 held by UCL Business plc.
Additional information
Supplementary Information accompanies this paper on Eye website
Rights and permissions
About this article

Cite this article
Dart, J. The 2016 Bowman Lecture Conjunctival curses: scarring conjunctivitis 30 years on. Eye 31, 301–332 (2017). https://doi.org/10.1038/eye.2016.284
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2016.284
This article is cited by
-
Association of topical glaucoma medications with lacrimal drainage obstruction and eyelid malposition
Eye (2023)
-
Erste Ergebnisse des Deutschen okulären Pemphigoid-Registers
Die Ophthalmologie (2023)
-
Klinik des okulären Schleimhautpemphigoids
Die Ophthalmologie (2023)
-
Okuläres Schleimhautpemphigoid
Die Ophthalmologie (2023)
-
Immunpathogenese des Schleimhautpemphigoids
Die Ophthalmologie (2023)
