
Abstract
Purpose
To determine the genetic basis of early onset autosomal recessive Best vitelliform macular dystrophy (arBVMD) in a family with three affected children.
Design
Clinical and family-based genetic study.
Methods
Seven subjects making up a family with three children affected by Best vitelliform macular dystrophy were studied. Standard ophthalmic exam with dilated ophthalmoscopy and imaging were performed in each individual. The eleven exons of BEST1were directly sequenced.
Results
All three affected children have the clinical characteristic features of Best vitelliform macular dystrophy: large macular vitelliform lesions, scattered vitelliform lesions along the arcades and in the peripheral retina, and an accumulation of serous retinal fluid. A novel compound heterozygous mutation in the BEST1gene was found in the three affected individuals (L41P and I201T). The unaffected parents and children only harbor one heterozygous mutation.
Conclusion
arBVMD can be caused by the compound heterozygous mutation L41P and I201T in the BEST1gene.
Similar content being viewed by others


Introduction
Autosomal dominant Best vitelliform macular dystrophy (BVMD) is one of the most common retinal dystrophies. The disease is characterized clinically by deposition of yellowish material in the retinal pigment epithelium (RPE) and subretinal space. This accumulated material forms the characteristic egg yolk-like appearance in the macula. While the autosomal dominant pattern of inheritance has been long observed, an autosomal recessive pattern has also been reported recently.1 Homozygous or compound mutations in the BEST1 gene can cause autosomal recessive BVMD (arBVMD).1, 2, 3, 4, 5, 6
BEST1 is located on chromosome 11q13 and encodes the bestrophin-1 protein that localizes to the RPE.1, 7, 8 The exact function of the bestrophin protein is not completely understood. However, it has been suggested that bestrophin-1 acts as either a chloride channel or a modulator of calcium channels.6, 8, 9, 10
To date, several mutations in arBVMD cases have been reported.1, 2, 3, 5, 11, 12 These include homozygous and combinations of nonsense or missense mutations. In all of these cases, patients presented with central vision loss, characteristic retinopathy, absence of electro-oculogram light rise, and a reduced electroretinogram.1 In this report, we present a novel compound heterozygous mutation in a family with three affected children.
Materials and methods
Participants and clinical examinations
The family being studied consisted of seven individuals, including two parents and five children. Three of the children complained of visual impairment. The parents and two of the children denied any visual symptoms. Both of the parents, the three affected children, and two non-affected children had their blood drawn for genetic analysis.
Each family member received a complete ophthalmic examination. Exams included best-corrected visual acuity measurements and applanation tonometry for intraocular pressure measurements. Slit lamp exams included fundus biomicroscopy. In addition, fundus photography and spectral domain optical coherence tomography were completed.
DNA sequencing
Genomic DNA samples were extracted from peripheral blood leukocytes according to established protocols from seven of the family members. Direct DNA sequence analysis was completed for the BEST1 gene. The eleven exons of the BEST1 gene were amplified by PCR and sequenced using the Genetic Analyzer 3130 system (Applied Biosystems, Foster City, CA, USA). Primers used to amplify the exons in the BEST1 gene are presented (Table 1).
Results
Clinical examinations
The family under study consisted of seven individuals, including two non-consanguineous Caucasian parents and five children. Three of the five children were diagnosed with arBVMD at age 4.
Child 1 (II-1 in Figure 1a) is a twelve-year-old female with best-corrected visual acuities of 20/400 OD and 20/30 OS. Dilated ophthalmic exam is significant for macular lesions and smaller scattered vitelliform lesions in the periphery of the right eye, intraretinal cysts, and elevation and separation of the neurosensory retina from the RPE in the left eye (Figure 2).

Pedigree and the BEST1 sequencing results for a family affected by arBVMD. (a) Pedigree of the family affected by arBVMD. Father (I-1), mother (I-2), and the two children (II-2 and II-5) are normal; each one harbors only one mutation (L41P or I201T). The other three children (II-1, II-3, and II-4) are affected by arBVMD and have the compound mutation (L41P and I201T). (b) Sequencing data show that a T>C transition in exon 5 (arrow) causes the L41P mutation and (c) that a T>C transition in exon 2 (arrow) causes the I201T mutation.

(II-1) Fundus photographs (a, c) of both eyes significant for macular lesions surrounded by retinal elevation bilaterally. Multiple scattered vitelliform lesions are noted in the periphery outside the arcades. Optical coherence tomography images (b, d) are shown through the macular lesions. Significant distortion of the macular structure is noted with elevation and separation from the RPE and the presence of intraretinal cysts and subretinal fluid (c, d).
Child 3 (II-3 in Figure 1a) is a nine-year-old male with best-corrected visual acuities of 20/30 OD and 20/40 OS. Dilated ophthalmic exam is significant for scattered vitelliform lesions, subretinal deposits, and subretinal fluid in the macula of the right eye (Figure 3).

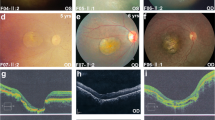
(II-3) Fundus photographs (a, c) of both eyes significant for many small, scattered vitelliform lesions in the macula and larger vitelliform lesions along the arcades and in the periphery. Additionally, there are macular vitelliform lesions in both eyes with serous retinal fluid in the macula of the right eye. Optical coherence tomography images (b, d) through the macular and peripheral lesions are also shown. The peripheral lesion in the right eye (a, b) is located nasal to the macula (white arrow upper left panel), and the retina is also separated from the RPE. The scan through the macular lesion (d) shows elevation of the macular area with intraretinal fluid.
Child 4 (II-4 in Figure 1a) is a five-year-old female with best-corrected visual acuities of 20/40 OD and 20/30 OS. Dilated ophthalmic exam is significant for bilateral yellowish, subretinal macular lesions with the lesion in the right eye extending outside the macular area down to the inferior arcade. There are some small deposits noted in the left macula and serous retinal fluid in both eyes.
The parents (I-1 and I-2) and the other two unaffected children (II-2 and II-5) have no clinical signs of disease (Figure 4).

(I-2) Fundus photographs (a, c) and optical coherence tomography images (b, d) of both eyes reveal normal anatomy and no signs of retinal disease.
Genetic analysis
The family history is consistent with an autosomal recessive inheritance pattern (Figure 1a). We found the unaffected father to have a T>C transition (c.122T>C) in exon 5, resulting in a L41P substitution (Figure 1b). The unaffected mother, however, was found to have a different variation, consisting of a T>C transition (c.602T>C) in exon 2, resulting in an I201T substitution (Figure 1c). The three affected children harbor both of these variations while the two unaffected children carry only one of the heterozygous mutations. Table 2 summarizes the genetic analysis results for each family member. These two changes were absent in 1000 normal control chromosomes.
Discussion
We report a family with an autosomal recessive BVMD that resulted from compound heterozygous BEST1 mutations.1 In these cases, the unaffected family members carry only one of the heterozygous mutations. While over 100 different BEST1 mutations have been described in families affected by Best disease,13, 14, 15, 16, 17 only a few combinations of compound heterozygous mutations in arBVMD have been reported.1, 2, 3, 4, 5, 11
In our study, the affected children were found to have a combination of two heterozygous mutations: c.122T>C (L41P) and c. 602T>C (I201T). The L41P mutation has been previously implicated in arBVMD as a compound heterozygous mutation with P152A in one case, and with R141H in a second case.1 The I201T mutation has also been implicated in arBVMD. To date, three individual cases of a compound heterozygous mutation with I201T have been reported: I201T with P152A, I201T with IVS 7+4G>A, and I201T with the deletion Phe281del3CAGTTC.1 We have shown for the first time that the combination of I201T with L41P can also lead to arBVMD.
In vitro studies using HEK293 cells have shown that co-transfection of the two mutations observed in the compound heterozygous state in arBVMD patients essentially abolished chloride channel activity. However, when six arBVMD mutations were coexpressed with wild-type bestrophin-1, none of the combinations suppressed the wild-type chloride channel activity.1 Therefore, it is suggested that the autosomal recessive phenotype only manifests when the bestrophin-1 activity drops below a functional threshold.1, 18 The mutations that make up the autosomal recessive phenotype do not cause enough structural change in the protein to bring its function below threshold alone in the carrier. They must be combined with other mutations to affect protein function.
The phenotype of BVMD varies with each patient and the stage of the disease. Initial stages are characterized by a circular or horizontally elliptical vitelliform lesion centered around the fovea and ranging in diameter from 0.5 disk diameter to 2 disk diameters, simulating the appearance of an egg yolk.19 Over the years, the material in the subretinal space may become less homogenous, giving rise to what is known as the ‘scrambled-egg’ stage, or it may gravitate inferiorly, forming the ‘pseudohypopyon’ stage.20 Previous publications have discussed that the clinical presentation of cases of compound arBVMD are noticeably different from the autosomal dominant form.1, 11, 13 The most-reported distinguishing feature of arBVMD is extrafoveal and extramacular subretinal deposits.
Additionally, patients with BVMD are at increased risk for choroidal neovascularization. These membranes are often difficult to detect owing to the overlying vitelliform lesions.3 There have been several reports of favorable outcomes with the use of anti-VEGF therapy.21, 22 This has turned out to be consistent with our study, where the patients’ sub- or intraretinal fluid has resolved from injections of bevacizumab.
Our finding adds to the spectrum of clinical presentations caused by BEST1 mutations. Understanding the genetics of the bestrophinopathies and the cause of different forms of the disease will allow for the development of potential gene therapy regimens and new diagnostic modalities.

References
Burgess R, Millar ID, Leroy BP, Urquhart JE, Fearon IM, De Baere E et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet 2008; 1: 19–31.
Kinnick TR, Mullins RF, Dev S, Leys M, Mackey DA, Kay CN et al. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients. Retina 2011; 3: 581–595.
Iannaccone A, Kerr NC, Kinnick TR, Calzada JI, Stone EM . Autosomal recessive best vitelliform macular dystrophy: report of a family and management of early-onset neovascular complications. Arch Ophthalmol 2011; 2: 211–217.
Davidson AE, Sergouniotis PI, Burgess-Mullan R, Hart-Holden N, Low S, Foster PJ et al. A synonymous codon variant in two patients with autosomal recessive bestrophinopathy alters in vitro splicing of BEST1. Mol Vis 2010; 16: 2916–2922.
Gerth C, Zawadzki RJ, Werner JS, Heon E . Detailed analysis of retinal function and morphology in a patient with autosomal recessive bestrophinopathy (ARB). Doc Ophthalmol 2009; 3: 239–246.
Hartzell C, Qu Z, Putzier I, Artinian L, Chien LT, Cui Y . Looking chloride channels straight in the eye: bestrophins, lipofuscinosis, and retinal degeneration. Physiology (Bethesda) 2005; 20: 292–302.
Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet 1998; 3: 241–247.
Sun H, Tsunenari T, Yau KW, Nathans J . The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA 2002; 6: 4008–4013.
Tsunenari T, Sun H, Williams J, Cahill H, Smallwood P, Yau KW et al. Structure-function analysis of the bestrophin family of anion channels. J Biol Chem 2003; 42: 41114–41125.
Rosenthal R, Bakall B, Kinnick T, Peachey N, Wimmers S, Wadelius C et al. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J 2006; 1: 178–180.
Schatz P, Klar J, Andreasson S, Ponjavic V, Dahl N . Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet 2006; 2: 51–56.
Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI . The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res 2009; 3: 187–205.
Lotery AJ, Munier FL, Fishman GA, Weleber RG, Jacobson SG, Affatigato LM et al. Allelic variation in the VMD2 gene in best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci 2000; 6: 1291–1296.
Kramer F, Mohr N, Kellner U, Rudolph G, Weber BH . Ten novel mutations in VMD2 associated with Best macular dystrophy (BMD). Hum Mutat 2003; 5: 418.
Caldwell GM, Kakuk LE, Griesinger IB, Simpson SA, Nowak NJ, Small KW et al. Bestrophin gene mutations in patients with Best vitelliform macular dystrophy. Genomics 1999; 1: 98–101.
Downs K, Zacks DN, Caruso R, Karoukis AJ, Branham K, Yashar BM et al. Molecular testing for hereditary retinal disease as part of clinical care. Arch Ophthalmol 2007; 2: 252–258.
Palomba G, Rozzo C, Angius A, Pierrottet CO, Orzalesi N, Pirastu M . A novel spontaneous missense mutation in VMD2 gene is a cause of a best macular dystrophy sporadic case. Am J Ophthalmol 2000; 2: 260–262.
Davidson AE, Millar ID, Burgess-Mullan R, Maher GJ, Urquhart JE, Brown PD et al. Functional characterization of bestrophin-1 missense mutations associated with autosomal recessive bestrophinopathy. Invest Ophthalmol Vis Sci 2011; 6: 3730–3736.
Blodi CF, Stone EM . Best's vitelliform dystrophy. Ophthalmic Paediatr Genet 1990; 1: 49–59.
Yannuzzi LA . The Retinal Atlas. Saunders/Elsevier: Philadelphia, PA, 2010.
Querques G, Bocco MC, Soubrane G, Souied EH . Intravitreal ranibizumab (Lucentis) for choroidal neovascularization associated with vitelliform macular dystrophy. Acta Ophthalmol 2008; 6: 694–695.
Leu J, Schrage NF, Degenring RF . Choroidal neovascularisation secondary to Best's disease in a 13-year-old boy treated by intravitreal bevacizumab. Graefes Arch Clin Exp Ophthalmol 2007; 11: 1723–1725.
Acknowledgements
This study was approved by the Institutional Review Board of the University of California, San Diego. All subjects signed informed consent before participation in the study. We thank the family for their support. This study was supported by NEI/NIH grants, Research to Prevent Blindness, San Diego Clinical and Translational Research Institute 1TL1RR031979-01, and the VA Merit Award.
Author contributions
Design of the study (LZ, SG and KZ); conduct of the study (LZ, SG, MK, JL, HD and GH); data collection (SG, RC, JL, CL and IK); analysis and interpretation of the data (LZ, SG, MK, JQ, JZ, IK, PXS and KZ); and writing of the manuscript (LZ, SG, MK, PXS, IK and KZ).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Access to data
The principal investigator, Dr Kang Zhang, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Rights and permissions
About this article
Cite this article
Zhao, L., Grob, S., Corey, R. et al. A novel compound heterozygous mutation in the BEST1 gene causes autosomal recessive Best vitelliform macular dystrophy. Eye 26, 866–871 (2012). https://doi.org/10.1038/eye.2012.27
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2012.27
