
Abstract
Purpose
Mutations in the 1A-subunit of the brain P/Q-type calcium channel gene CACNA1Aare responsible for spinocerebellar ataxia type 6 (SCA6), familial haemiplegic migraine (FHM) and episodic ataxia type 2 (EA2). Considerable clinical and genetic overlap exists between these 3 allelic disorders. Clinical findings are varied and may include nystagmus.
Objective
To study the clinical phenotype and identify a causative mutation in a family who presented when the youngest member was diagnosed with apparent isolated congenital nystagmus (age 3 months).
Patients and Methods
8 patients from one family underwent detailed clinical phenotyping comprising; ophthalmic and neurological examination, nystagmology, electrodiagnostic tests and brain imaging. The CACNA1Agene was screened for mutations by direct sequencing in one patient. Co-segregation of the disease and an identified sequence variation was shown using direct sequencing.
Results
Phenotyping revealed isolated atypical nystagmus in 4 family members and nystagmus in addition to late onset ataxia in 1 family member. Direct sequencing of the CACNA1Agene identified a novel missense mutation; (c.4110T>G p.Phe1370Leu (NM_000068.3)).
Conclusions
We have shown that a mutation in the intracellular domain between s4 and s5 of repeat 3 can cause atypical nystagmus/cerebellar phenotypes, including isolated nystagmus in an infant. We also illustrate the necessity for detailed examination of relatives in cases of apparent isolated congenital nystagmus.
Similar content being viewed by others

Introduction
Nystagmus before six months of age can be defined as early onset nystagmus.1 It may be divided into three categories: sensory defect nystagmus (SDN), congenital idiopathic nystagmus (CIN) and neurological nystagmus (NN), which is associated with neurological disease.1
Nystagmus can be a clinical feature in three allelic disorders namely spinocerebellar ataxia type 6 (SCA6, MIM 183086), familial haemiplegic migraine (FHM, MIM 141500) and episodic ataxia type 2 (EA2, MIM 108500). SCA6 is characterised by adult-onset, slowly progressive cerebellar ataxia, dysarthria, nystagmus, and impaired sensations of vibration and proprioception.2 FHM is characterised by migraine attacks preceded by symptoms, such as unilateral limb paresis or paralysis, paraesthesias, dysphasia, and interictal cerebellar signs, which are reported in about 50% of patients.3 EA2 is characterised by a complex and highly variable phenotype, widely overlapping that of SCA6.4 The main features are episodes of vertigo or ataxia of variable frequency and duration, a permanent cerebellar deficit of variable severity, and a cerebellar atrophy typically starting from the anterior portion of the vermis.5 Significant clinical overlap exists between these three disorders.6
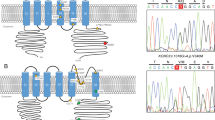
These disorders can all be caused by mutations in the CACNA1A gene (OMIM 601011). CACNA1A codes for the Cav2.1 α-1A subunit of P/Q-type voltage gated Ca2+ channels (Ca(V)2.1) expressed in the brain, and particularly in cerebellar Purkinje and granule cells, as well as in neuromuscular junctions.7, 8 The protein has four domains or repeats, each formed by six transmembrane hydrophobic segments. Typically, point mutations in this gene are responsible for EA2 and FHM (see Figure 3), whereas small expansions of a CAG repeat characterise SCA6.

Published mutations in of the CACNA1A gene within the Ca(V)2.1 protein product. Non-truncating (red dots) and truncating (orange dots) mutations in subjects with EA2 are presented with mutations from patients with FHM (green dots) and the c.4110T>G mutation identified in this work (red circle with yellow centre).
Typically, nystagmus in these disorders comprises vertical jerk (downbeat, upbeat), gaze evoked, pendular, periodic alternating, or rebound nystagmus.9, 10 Smooth pursuit is typically saccadic, optokinetic nystagmus (OKN) absent or of low gain, and saccades may be dysmetric.9, 10 Here, we present a family with a novel CACNA1A mutation with presentation as an isolated case of atypical CIN in an infant.
Materials and methods
Clinical phenotyping methods
Eight individuals in a single pedigree (Figure 1) were interviewed, and detailed clinical examination was completed, including LogMAR visual acuity, slit-lamp ophthalmic examination, full orthoptic assessment, and clinical neurological examination. Subjects in whom nystagmus was found also had eye movement recordings using a Skalar IRIS IR Light Eye Tracker equipment (Cambridge Research Systems Ltd., Rochester UK, www.crsltd.com) as previously described by us.11 Subject IV:1 (infant) did not have eye movement recordings because of her age. Three patients also had extensive electrophysiological examinations. The standardised electroretinograms (ERGs) of the International Society for Clinical Electrophysiology of Vision (www.iscev.org) and visual-evoked potential (VEP) recordings were recorded in three female subjects (III:1, II:1, and IV:1). Magnetic resonance brain scans were performed and sagittal t1 and axial dual echo images were obtained in individuals II:1 and IV:1.

Cosegregation of a CACNA1A variation (c.4110T>G) in a pedigree with atypical hereditary nystagmus. A pedigree diagram is presented with subject IDs, age, and genotyping. Variant sequence shows a heterozygote peak at position 3, and wild-type sequence shows a single red peak corresponding to a thymine base.
Molecular genetic methods
Genomic DNA was isolated from venous blood by standard techniques.
The complete CACNA1A gene (exons 1–47) (NM_000068.3) was screened in III:I by direct sequencing. Cosegregation of a sequence variation in the pedigree was performed by direct sequencing in all eight family members. A total of 150 control subjects were subsequently screened for this variation by high-resolution melt (HRM) analysis on a Rotor-Gene™ 6000 realtime thermocycler (Corbett Life Science Ltd., Brisbane, Australia). Assays were performed according to the manufacturer's instructions and primer sequences are available on request.
Results
Clinical
Nystagmus was identified in five of the eight subjects examined (Figure 1). Subject; II:1, a 54-year-old female, also described a mild ataxia starting at the age of 35 years with slow progression thereafter. She had noticed fluctuations in her ataxia and dysarthria but without typical episodes of EA2. She had occasional migrainous headaches. Examination showed a broad-based gait, no dysarthria, moderate finger–nose ataxia, mild shin–heel ataxia, and reduction in accuracy of alternating hand movements. No weakness was found. Tendon reflexes were normal; she had flexor plantar responses and no sensory loss. Interestingly, she had no response to oral acetazolamide treatment for 3 weeks, and this was subsequently discontinued. All the other family members had no clinical symptoms or signs of neurological disease besides nystagmus. Subject III:3 was seen 2 years after the clinical phenotyping in this family (now age 32 years), and at that stage had developed a mild cerebellar ataxia with no other clinical signs besides nystagmus. MR brain scanning in subject IV:1 identified no anomalies and a normal cerebellum, although a scan of subject II:1, when she was 50 years old, had shown a ‘borderline,’ small cerebellar vermis. VEP and ERG recordings were normal for all examined subjects.
Detailed nystagmology revealed a similar pattern in all subjects with downbeat nystagmus in primary gaze, average nystagmus amplitude of 2.5 degrees and frequency of 2.5 beats/s, gaze-evoked nystagmus in other positions of gaze, low gain OKN, saccadic pursuit, and dysmetric saccades. Exceptions were: steady fixation (IV:3) and left-beating nystagmus (III:3) in primary gaze. Rebound nystagmus was only seen in III:1. All waveforms exhibited linear or decelerating slow phases, no accelerating slow phases were seen in any waveform.
Molecular genetic results
Direct sequencing of the complete CACNA1A gene for subject III:1 identified a variant; Chr19(NCBI 36):g. 13233416T>G, which corresponds to c.4110T>G p.Phe1370Leu (exon 26) in the NM_000068.3 transcript. No other pathogenic mutations were identified in this gene. This variant was not previously described and not detected in 150 unrelated control subjects (300 chromosomes) by HRM analysis. To determine the pathogenicity of this variant, segregation analysis was performed. Direct sequencing of the c.4110T>G variant in subjects III:1–8 showed that the variant cosegregates with the disease phenotype (Figure 1).
Discussion
We describe a novel mutation in the CACNA1A gene in a family who presented with apparent isolated atypical congenital nystagmus in an infant.
Cosegregation with the disease phenotype and absence from 300 control chromosomes and 480 FHM/EA-2 probands provides good supportive evidence for the pathogenicity of the c. 4110T>G sequence change in this pedigree. Further support is given by the observation that this variation occurs at a position, which is highly conserved throughout evolution (Figure 2).

Conservation of the phenylalanine amino acid, which is altered by the CACNA1A c.4110T>G p.Phe1370Leu (NM_000068.3) variation. The phenylalanine amino acid (F) is surrounded by red box. Sequences are from the UNIPROT database (http://beta.uniprot.org/).
Some groups have suggested that the clinical phenotype associated with mutations in this gene is predicted by the class/location of the mutation.5 Other groups disagree.12 However, the unusual clinical phenotype in this family may be due to the location of the mutation in part of the Ca(V)2.1 gene product. Only one other mutation has been identified in this region (C1369Y mutation) in a pedigree with FHM.13 However, this mutation was present in only three of the four affected family members, and therefore did not fully cosegregate with the disease phenotype.
Using a protein sequence and structure database (http://beta.uniprot.org/uniprot/O00555), the c.4110T>G p.Phe1370Leu (NM_000068.3) variation identified in this work is predicted to be located within a small 19-bp topological domain of the Ca(V)2.1 gene product. This domain is positioned within the cytoplasm between segments 4 and 5 of the repeat domain 3 (Figure 3). Interestingly, the family described in this work presented with a number of features, which are unusual for the spectrum of diagnoses spanning EA2, SCA6, and FHM. They presented because of isolated nystagmus in a 3-month-old baby (IV:1), which has not been reported previously. In EA2, interictal nystagmus tends to occur at the same time or later than the onset of other cerebellar symptoms. Usually, it is evident after the development of cerebellar vermis atrophy, which this child did not have. Only one of the eight subjects in this pedigree described migrainous headaches and none describe episodes of haemiplegia. Indeed, all but the 3-month-old proband with nystagmus and two adults with the onset of subtle ataxia at the age of 32 and 35 years were completely asymptomatic and had no other signs of cerebellar dysfunction except for subtle nystagmus at the time of examination. This suggests that for this family, the nystagmus is early in onset and ataxia only develops at around 30–35 years of age.
The nystagmus waveforms in this pedigree are quite typical of cerebellar nystagmus. Unusually, three of the five affected subjects were not aware of their nystagmus before our examination. One possible explanation for this is the low intensity (frequency x amplitude) of the waveform in these subjects and the complete absence of nystagmus in the primary position in one subject. It also may suggest that the nystagmus had been present since birth as oscillopsia is far more common in acquired than congenital nystagmus.
This family initially presented with a diagnosis of atypical CIN in IV:1 at 3 months old following MR brain, VEP, ERG, and full examination. This emphasises the importance of ophthalmic examination of relatives in cases of apparent sporadic CIN, particularly if the waveform is atypical. It also illustrates that a missense mutation, typically associated with FHM1, presented with early manifestation of congenital downbeat nystagmus and late onset slowly progressive ataxia that are classic signs in SCA6, but not prominent migraine symptoms further blurring the three allelic disorders EA2, SCA6, and FHM1.
References
Casteels I, Harris CM, Shawkat F, Taylor D . Nystagmus in infancy. Br J Ophthalmol 1992; 76: 434–437.
Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet 1997; 15: 62–69.
Headache Classification Subcommittee of the International Headache Society. The International Classification of Headache Disorders: 2nd edition. Cephalalgia 2004; 24 (Suppl 1): 9–160.
Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW . Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 2007; 130: 2484–2493.
Mantuano E, Veneziano L, Spadaro M, Giunti P, Guida S, Leggio MG et al. Clusters of non-truncating mutations of P/Q type Ca2+ channel subunit Ca(v)2.1 causing episodic ataxia 2. J Med Genet 2004; 41: e82.
Mantuano E, Veneziano L, Jodice C, Frontali M . Spinocerebellar ataxia type 6 and episodic ataxia type 2: differences and similarities between two allelic disorders. Cytogenet Genome Res 2003; 100: 147–153.
Volsen SG, Day NC, McCormack AL, Smith W, Craig PJ, Beattie R et al. The expression of neuronal voltage-dependent calcium channels in human cerebellum. Brain Res Mol Brain Res 1995; 34: 271–282.
Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP et al. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci 1995; 15: 6403–6418.
Rett D . Gaze-evoked nystagmus: a case report and literature review. Optometry 2007; 78: 460–464.
Durig JS, Jen JC, Demer JL . Ocular motility in genetically defined autosomal dominant cerebellar ataxia. Am J Ophthalmol 2002; 133: 718–721.
Self JE, Shawkat F, Malpas CT, Thomas NS, Harris CM, Hodgkins PR et al. Allelic Variation of the FRMD7 Gene in Congenital Idiopathic Nystagmus. Arch Ophthalmol 2007; 125: 1255–1263.
Jen J, Kim GW, Baloh RW . Clinical spectrum of episodic ataxia type 2. Neurology 2004; 62: 17–22.
Thomsen LL, Kirchmann M, Bjornsson A, Stefansson H, Jensen RM, Fasquel AC et al. The genetic spectrum of a population-based sample of familial hemiplegic migraine. Brain 2007; 130: 346–356.
Acknowledgements
We thank the family for their participation and the Nystagmus Network and MRC for funding. Mr Self holds an MRC clinical training fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Self, J., Mercer, C., Boon, E. et al. Infantile nystagmus and late onset ataxia associated with a CACNA1A mutation in the intracellular loop between s4 and s5 of domain 3. Eye 23, 2251–2255 (2009). https://doi.org/10.1038/eye.2008.389
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2008.389
Keywords
This article is cited by
-
Analysis of Fibroblast Growth Factor 14 (FGF14) structural variants reveals the genetic basis of the early onset nystagmus locus NYS4 and variable ataxia
European Journal of Human Genetics (2023)
-
Supranuclear eye movements and nystagmus in children: A review of the literature and guide to clinical examination, interpretation of findings and age-appropriate norms
Eye (2019)
-
A small gene sequencing panel realises a high diagnostic rate in patients with congenital nystagmus following basic phenotyping
Scientific Reports (2019)
-
Episodic ataxia type 2: phenotype characteristics of a novel CACNA1A mutation and review of the literature
Journal of Neurology (2014)
