
Abstract
Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is a tumor suppressor. Although it is well known to have various physiological roles in cancer, its inhibitory effect on inflammation remains poorly understood. In the present study, a human PTEN gene was fused with PEP-1 peptide in a bacterial expression vector to produce a genetic in-frame PEP-1-PTEN fusion protein. The expressed and purified PEP-1-PTEN fusion protein were transduced efficiently into macrophage Raw 264.7 cells in a time- and dose-dependent manner when added exogenously in culture media. Once inside the cells, the transduced PEP-1-PTEN protein was stable for 24 h. Transduced PEP-1-PTEN fusion protein inhibited the LPS-induced cyclooxygenase 2 (COX-2) and iNOS expression levels in a dose-dependent manner. Furthermore, transduced PEP-1-PTEN fusion protein inhibited the activation of NF-κB induced by LPS. These results suggest that the PEP-1-PTEN fusion protein can be used in protein therapy for inflammatory disorders.
Similar content being viewed by others


Introduction
Phosphatase and tensin homologue deleted on chromosome 10 (PTEN), also named MMAC-1 (mutated multiple advanced cancers) or TEP-1 (TGF-β-regulated and epithelial cell-enriched phosphatase) was identified as a new tumor suppressor gene involved in a wide variety of human cancers located at 10q23 (Li et al., 1997; Steck et al., 1997). PTEN is well known to play a key role in suppressing cancer, cell migration, survival and apoptosis (Yamada and Araki, 2001). PTEN is a major negative regulator of the phosphatidylinositol 3-kinase (PI3K) and serine/theronine protein kinase (Akt) signaling pathway by catalyzing degradation of the phosphatidylinositol-3,4,5-triphosphate (PIP3) to PI-4,5-diphosphate (Vazquez and Sellers, 2000).
Prostaglandins (PGs) are potent proinflammatory mediators derived form arachidonic acid metabolism by cyclooxygenase (COXs), and play an important role in modulating a number of pathophysiological conditions, including inflammatory and allergic immune response (Tilly et al., 2001). The two isoforms of COX enzymes have been well studied. COX-1 is constitutively expressed and plays an important role in maintaining the normal physiological function of cells. COX-2 is markedly induced by a number of stimuli including cytokines during the inflammatory response (Smith and Dewitt, 1990; Carey et al., 2003; Vancheri et al., 2004).
LPS is the main component of endotoxin and is formed by a phosphoglycolipid that is covalently linked to a hydrophilic heteropolysaccharide (Rietschel et al., 1994). LPS arrests macrophage proliferation and activates them to produce proinflammatory factors, which play important roles in the immune response (Adams and Hamilton, 1984; Morrison and Ryan, 1987). Eosinophils act as effectors in the inflammatory reactions of allergic diseases such as asthma, allergic rhinitis, and atopic dermatitis (AD), as well as in the chronic development of allergic inflammation (Fugihara et al., 2002; Gleich, 2000; Wong et al., 2002). Kwak et al. (2003) demonstrated that administration of an adenovirus gene transfer vector expressing a PTEN cDNA inhibitors reduced the inflammation and airway hyper-responsiveness in a murine model of allergic asthma. Although there has been increased interest in the role of PTEN in cellular function particularly in inflammations, the underlying mechanisms still need to be established.
Several small regions of proteins, called protein transduction domains (PTDs), have been developed to allow the delivery of exogenous protein into living cells. Up to the present, many researchers have demonstrated the successful delivery of full-length Tat fusion proteins by protein transduction technology (Wadia and Dowdy, 2002). We successfully transduced Tat-SOD directly into insulin-producing RINm-5F and pancreatic islet cells and transduced Tat-SOD by increased radical scavenger activity in the pancreas (Eum et al., 2002, 2004a). Recently, we showed that Tat-pyridoxal kinase (Tat-PK) and Tat-pyridoxal oxidase (Tat-PO) fusion protein were efficiently transduced into PC12 cells and catalytically active in the cells (Kim et al., 2005, 2006). Also, we transduced PEP-1-SOD and PEP-1-CCS into neuronal cells and across the blood-brain barrier which efficiently protected against ischemic insults. Also, Tat-α-synuclein fusion protein protects against oxidative stress in vitro and in vivo (Eum et al., 2004b; Choi et al., 2005, 2006a,b).
In the present study, we designed the PEP-1-PTEN fusion protein by genetic in-frame transduction and showed that the PEP-1-PTEN fusion protein can be directly transduced into macrophage Raw 264.7 cells, as well as inhibit levels of iNOS and COX-2 mRNA and protein in LPS-induced cells. Therefore, we suggest that PEP-1-PTEN fusion protein could be useful as a potential therapeutic agent for inflammatory diseases.
Materials and Methods
Materials
Restriction endonuclease and T4 DNA ligase were purchased from Promega Co. (Madison, WI). Oligonucleotides were synthesized from Gibco BRL custom primers (Grand Island, NY). Ni2+-nitrilotriacetic acid sepharose superflow was purchased from Qiagen (Valencia, CA). Isopropyl-β-D-thiogalactoside (IPTG) was obtained from Duchefa Co. (Haarlem, Netherlands). Plasmid pET-15b and Escherichia coli strain BL21 (DE3) were obtained from Novagen (Hilden, Germany). A human PTEN cDNA fragment was isolated using the PCR technique. Primary antibodies against histidine, COX-2, actin (Santa Cruz, CA) and phosphor-IκBα, total IκBα (Cell signaling Technology, Beverly, MA) were obtained commercially. All other chemicals and reagents were of the highest analytical grade available.
Cloning and purification of PEP-1-PTEN fusion proteins
The PEP-1 expression vector was constructed to express the PEP-1 peptides (KETWWETWWTEWSQPKKKRKV) as a fusion with human PTEN. First, two oligonucleotides (top strand) 5'-TATGAAAGAAACCTGGTGGGAAACCTGGTGGA CCGAATGGTCTCAGCCGAAAAAAAAACGTAAA GTGC-3' and (bottom strand) 5'-TCGABCACTTTACGTTTTTTTTTCGGCTGAGACCATTCGGTC CACCAGGTTTCCCACCAGGTTTCTTTCC-3' were synthesized and annealed to generate a double-stranded oligonucleotide encoding the PEP-1 peptides. The double-stranded oligonucleotide was directly ligated into a NdeI-XhoI digested pET-15b vector. Second, on the basis of the cDNA sequence of human PTEN, two primers were synthesized. The sense primer, 5'-CTCGAGATGGCAGCCATCATCAAAGAGATC-3' contains an XhoI site, and the antisense primer, 5'-GGATCCTCAGACTTTTGTAATTTGTGTATG-3', contains a BamHI restriction site. The resulting PCR products were digested with XhoI and BamHI, eluted (Invitek, Berlin, Germany), ligated into a TA-cloning vector (Promega, Madison, WI) and a pPEP-1 vector using T4 DNA ligase (Takaka, Otsu, Shiga, Japan), and cloned in E. coli BL21 (DE3) cells.
To produce the PEP-1-PTEN fusion protein, the plasmid was transformed into E. coli BL21 cells. The transformed bacterial cells were grown in 100 ml of LB media at 37℃ to a D600 value of 0.5-1.0 and induced with 0.5 mM IPTG at 37℃ for 3-4 h. Harvested cells were lysed by sonication at 4℃ in a binding buffer containing 5 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl (pH 7.9). After centrifugation, the supernatant was immediately loaded onto a Ni2+-nitrilotriacetic acid Sepharose affinity column (Qiagen, Valencia, CA). After the column was washed with 10 volumes of a binding buffer and 6 volumes of a washing buffer (35 mM imidazole, 500 mM NaCl, and 20 mM Tris-HCl, pH 7.9), the fusion protein was eluted with an eluting buffer (0.5 M imidazole, 500 mM NaCl, 20 mM Tris-HCl, pH 7.9). The fusion proteins were combined and the salts were removed using PD10 column chromatography (Amersham, Braunschweig, Germany). Then, the purified PEP-1-PTEN fusion proteins were further purified by chromatography on Superose 6 FPLC column. The protein concentration was estimated by the Bradford procedure using BSA as a standard (Bradford, 1976).
Transduction of PEP-1-PTEN into macrophage Raw 264.7 cell
The murine macrophage Raw 264.7 cells were cultured in DMEM containing 20 mM HEPES/NaOH (pH 7.4), 5 mM NaHCO3, 10% FBS and antibiotics (100 μg/ml streptomycin, 100 U/ml penicillin) at 37℃ under a humidified condition of 95% air and 5% CO2. For the transduction of PEP-1-PTEN, macrophage Raw 264.7 cells were grown to confluence on a 6-well plate. The culture medium was then replaced with fresh solution. After Raw 264.7 cells were treated with various concentrations of PEP-1-PTEN for 1 h, the cells were then treated with trypsin-EDTA and washed with PBS. Thereafter Raw 264.7 cells were harvested for the preparation of cell extracts to perform Western blot analysis.
The intracellular stability of transduced PEP-1-PTEN fusion protein was estimated by treating: after Raw 264.7 cells were treated with 3 μM native PEP-1-PTEN for 1 h, after which cells were washed and changed with a fresh culture medium to remove the PEP-1-PTEN that was not transduced. Cells were then further incubated for 48 h, followed by preparations of cell extracts for Western blot analysis.
Western blot analysis
The transduced PEP-1-PTEN proteins on the polyacrylamide gel were electrophoretically transferred to a nitrocellulose membrane. The membrane was blocked in 5% nonfat milk in Tris-buffered saline (TBS; 20 mM Tris, 0.2 M NaCl, pH 7.5) containing 0.05% Tween-20 (TBST) for 2 h and was then incubated for 1 h at room temperature with an anti-histidine antibody (Santa Cruz Biotechnology, Santa Cruz, CA; dilution 1:400) in TBST. After washing, the membrane was incubated for 1 h with a proper secondary antibody conjugated to HRP diluted 1:10,000 in TBST. The membrane was incubated with a chemiluminescent substrate and exposed to Hyperfilm ECL.
Determination of COX-2 protein expression
The macrophage Raw 264.7 cells were cultured in 6-well plates. The cells were washed with fresh medium and stimulated with 1 μg/ml LPS for 24 h. Then, the expression of COX-2 protein levels were determined by Western blotting using anti-COX-2 antibody.
Determination of NO production
NO was measured as its stable oxidative metabolite, nitrite, as described in (Misko et al., 1993). 100 μl of the culture medium was mixed with an equal volume of Griess reagent (0.1% naphthylethylenediamine dihydrochloride and 1% sulfanilamide in 5% phosphoric acid). The absorbance at 550 nm was measured, and the nitrite concentration was determined using a curve calibrated on sodium nitrite standards.
RT-PCR analysis
Total RNA was isolated from Raw 264.7 cells using a Trizol reagent kit (Invitrogen, Gaithersburg, MD) according to the manufacturer's instructions (Zhang et al., 2005). The RNA (2 μg) was reversibly transcribed with 10,000 U of reverse transcriptase and 0.5 μg/μl oligo-(dT) primer. PCR amplification of cDAN aliquots was performed with the following sense and antisense primers: COX-2 antisense, 5'-TGGACGAGGTTTTTCCACCAG-3'; sense, 5'-CAAAGGCCTCCATTGACCAGA3'; beta-actin antisense, 5'-GGACAGTGAGGCCAGGATGG-3'; sense, 5'-AGTGTGACG TTGACATCCGTAAAGA-3'; iNOS antisense, 5'-CTGTCAGAGCCTCGTGGCTTT-3'; sense, 5'-ATGGCTCGGGATGTGGCTAC-3'. PCR was performed in 50 μl of 10 mM Tris-HCl (pH 8.3), 25 mM MgCl2, 10 mM dNTP, 100 U of Taq DNA polymerase, and 0.1 μM of each primer and was terminated by heating at 70℃ for 15 min. PCR products were resolved on a 1% agarose gel and visualized with UV light in ethidium bromide.
Electrophoretic mobility shift assay (EMSA)
Raw 264.7 cells were treated with PEP-1-PTEN for 1 h, then nuclear extracts of the cells were prepared and analyzed for NF-κB binding activity by EMSA as described in (Song et al., 2007). An NF-κB consensus oligonucleotide was used in the EMSA. The complementary oligonucleotide was annealed and end-labeled with [γ-32P] ATP using T4 polynucleotide kinase. EMSA was performed in a total volume of 20 μl at 4℃. Five micrograms of nuclear extracts were equilibrated for 15 min in a binding buffer (10 mM Tris-HCl, pH 8.0, 75 mM KCl, 2.5 mM MgCl2, 0.1 mM EDTA, 10% glycerol, 0.25 mM DTT) and 1 μg of poly dl/Dc. 32P-labeled oligonucleotide probe (20,000 cpm) was then added and the reaction was incubated on ice for an additional 20 min. Bound and free DNA were then resolved by electrophoresis in a 6% native polyacrylamide gel in TBE buffer (89 mM Tris-HCl, 89 mM boric acid, and 2 mM EDTA).
Results
Construction and purification of PEP-1-PTEN fusion protein
To generate a cell-permeable expression vector, PEP-1-PTEN, a human PTEN cDNA was subcloned into the pET-15b plasmid that had been reconstructed to contain the PEP-1 peptide. The PEP-1-PTEN expression vector thus formed contained consecutive cDNA sequences encoding human PTEN, PEP-1 peptide (21 amino acids) and six histidine residues at the amino-terminus (Figure 1A). We also constructed the PTEN expression vector to produce control PTEN protein without PEP-1 transduction peptides.

Expression and purification of PEP-1-PTEN fusion protein. A schematic representation of the PEP-1-PTEN fusion protein containing 6His, PEP-1, and PTEN coding sequence (A). Protein extracts from cells and purified fusion proteins were analyzed by 10% SDS-PAGE (B, C) and subjected to Western blot analysis with an anti-rabbit polyhistidine antibody (D). Lanes in B are as follows: lane 1, non-induced; lane 2, induced PEP-1-PTEN; lane 3, induced PTEN. Lanes in C and D are as follows: lane 1, purified PEP-1-PTEN; lane 2, purified PTEN.
Following the induction of expression, the PEP-1-PTEN fusion proteins were purified. The fusion proteins were expressed in E. coli and the clarified cell extracts were loaded onto a Ni2+-nitrilotriacetic acid Sepharose affinity column under native conditions. A fusion protein containing fractions was combined and salts were removed using a PD10 column. A major single band was obtained by superpose 6 FPLC chromatography. The crude cell extracts obtained from E. coli and purified PEP-1-PTEN fusion proteins were electrophoresed in 10% SDS-PAGE (Figure 1B and C). The purified proteins were further confirmed by Western blot analysis using an anti-rabbit polyhistidine antibody. PEP-1-PTEN was detected at the corresponding bands in Figure 1C and D.
Transduction of PEP-1-PTEN into macrophage Raw 264.7 cells
To evaluate the transduction ability of PEP-1-PTEN fusion protein, we analyzed the transduction of PEP-1-PTEN proteins by adding them to Raw 264.7 cells culture media at 3 μM for various periods (15-60 min), and then analyzed the transduced protein levels by Western blotting. The intracellular concentration of transduced PEP-1-PTEN in cells was seen to gradually increase at 60 min (Figure 2A).

Transduction of PEP-1-PTEN fusion proteins. (A) 3 μM PEP-1-PTEN were added to the culture media of Raw 264.7 cells for 15-60 min, (B) 0.5-3 μM of PEP-1-PTEN were added to the culture media for 1 h, (C) cells were pre-treated with 3 μM PEP-1-PTEN incubated for 1-48 h, and analyzed by Western blot analysis. PTEN and β-actin were used as a control.
The dose-dependency of the transduction of PEP-1-PTEN fusion proteins was further analyzed. Various concentrations (0.5-3 μM) of PEP-1-PTEN proteins were added to Raw 264.7 cells in culture for 60 min, and the levels of transduced proteins were measured by Western blotting. As shown in Figure 2B, the levels of transduced proteins in Raw 264.7 cells increased in a concentration-dependent manner with the amount of fusion protein in the culture media. These results indicate that PEP-1-PTEN fusion proteins are transduced into Raw 264.7 cells in a dose- and time-dependent manner. However, control PTEN was not transduced into the cells.
The intracellular stability of transduced PEP-1-PTEN into Raw 264.7 cells is shown in Figure 2C. The PEP-1-PTEN fusion protein was added to the culture media of Raw 264.7 cells at a concentration of 3 μM for various time periods and the resulting levels of transduced protein were analyzed by Western blotting. The intracellular level of transduced PEP-1-PTEN fusion protein into cells was initially detected after 1 h, and then declined gradually over the period of observation. However, significant levels of transduced PTEN protein persisted in the cells for 24 h.
Effect of PEP-1-PTEN fusion protein on LPS-induced COX-2 expression and NO production
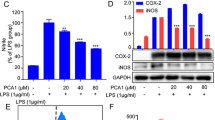
Macrophage plays crucial roles in the initiation and maintenance of inflammation. Since the level of COX-2 and NO is important in addressing the extent of inflammation, the effect of PEP-1-PTEN fusion protein on the inhibition of COX-2 expression and NO production was investigated. Raw 264.7 cells were incubated for 24 h with LPS (1 μg/ml) in the presence or absence of various concentrations (0.5-3 μM) of PEP-1-PTEN fusion protein. PEP-1-PTEN fusion protein suppressed LPS-induced COX-2 expression levels in Raw 264.7 cells in a dose-dependent manner (Figure 3).

Effect of transduced PEP-1-PTEN on LPS-induced COX-2 protein expression. Raw 264.7 cells were pretreated with the PEP-1-PTEN for 1 h before incubation with LPS (1 μg/ml) for 24 h. The cells were lysed and the lysates were analyzed by immunoblotting using an anti-COX-2 antibody.
The effect of PEP-1-PTEN fusion protein on NO production was examined in Raw 264.7 cells. Cells were stimulated with LPS (1 μg/ml) for 24 h in the presence or absence of various concentrations (0.5-3 μM) of PEP-1-PTEN fusion protein. After which cell culture media were collected and nitrite levels were determined. The exogenous PEP-1-PTEN fusion protein reduced NO production in a dose-dependent manner (Figure 4). However, the control PTEN protein did not suppress LPS-induced COX-2 expression levels in the same conditions and no significant cytotoxicity of PEP-1-PTEN fusion protein was determined in the cells (data not shown).

Effect of transduced PEP-1-PTEN on LPS-induced NO production. Raw 264.7 cells were pretreated with the PEP-1-PTEN for 1 h before incubation with LPS (1 μg/ml) for 24 h. Nitrite levels were measured in the culture media of LPS-stimulated cells by the Griess reaction. Each bar represents the mean ± SEM obtained from five experiments. *P < 0.05 and †P < 0.01 versus LPS alone. The statistical analysis was evaluated by Student's t-test.
We further examined the effects of PEP-1-PTEN on COX-2 and iNOS mRNA expression in LPS-induced cells by RT-PCR. As shown in Figure 5, post-treatment with PEP-1-PTEN fusion protein markedly inhibited LPS-induced mRNA expression of COX-2 and iNOS. These results suggest that the inhibition of COX-2 and iNOS mRNA expression by transduced PEP-1-PTEN fusion protein were responsible for the inhibition of COX-2 and NO production.

Inhibitory effect of PEP-1-PTEN on LPS-induced iNOS and COX-2 mRNA levels in Raw 264.7 cells. Raw 264.7 cells were pretreated with the PEP-1-PTEN for 1 h before incubation with LPS (1 μg/ml) for 12 h. Total RNA was extracted. iNOS and COX-2 mRNA was analyzed by RT-PCR using specific primers.
Effect of PEP-1-PTEN on LPS-induced NF-κB activation
As NF-κB plays critical role in the LPS-induced expression of iNOS and COX-2 in Raw 264.7 cells, we attempted to determine the effect of transduced PEP-1-PTEN on LPS-induced activation of NF-κB. Nuclear extracts from LPS-induced cells were analyzed using EMSA. As shown in Figure 6A, transduced PEP-1-PTEN fusion protein decreased in the LPS-induced DNA binding activity of NF-κB in a dose-dependent manner. Next, we examined the regulatory effects of PEP-1-PTEN fusion protein on the LPS-induced signal cascade of NF-κB activation such as IκBα phosphorylation and IκBα degradation. As shown in Figure 6B, PEP-1-PTEN fusion protein inhibited LPS-induced IκBα phosphyrylation and degradation in the cells.

Effect of transduced PEP-1-PTEN on LPS-induced activation of NF-κB in Raw 264.7 cells. Raw 264.7 cells were pretreated with the PEP-1-PTEN for 1 h before incubation with LPS (1 μg/ml) for 1 h and analyzed for NF-κB binding by EMSA (A). Phosphorylation and degradation of IκB-α were analyzed by Western blot analysis (B).
Discussion
It is well known that PTEN is a tumor suppressor gene that suppresses cell growth, inhibits cell migration, and induces apoptosis. It has been implicated in regulating cell survival signaling through the PI3K/Akt pathway (Maehama and Dixon, 1998; Myers et al., 1998; Cantley and Neel, 1999; Lu et al., 1999; Yamada and Araki, 2001).
Many inflammatory mediators attract and activate eosinophils via signal transduction pathways involving the enzyme PI3K (Dunzendorfer et al., 1998; Palframan et al., 1998; Tigani et al., 2001). Asthma is a chronic inflammatory disorder of the airways in which many cell types play a role. Eosinophil response appears to be a critical feature in asthma (Frigas and Gleich, 1986; Bousquet et al., 2000). It was reported that the PTEN protein expression and activity were decreased in OVA-induced asthma. They demonstrated that the administration of an adenovirus gene transfer vector expressing a PTEN cDNA or PI3K inhibitor reduced inflammation and airway hyperresponsiveness in a murine model of allergic asthma, and the inhibition of PI3K may be a good therapeutic strategy (Kwak et al., 2003). Several studies reported that the role of PTEN in immunity has been shown using PTEN-deficient mice. They have generated T cell-specific PTEN-deficient (tPtenflox/-) mice in which the T cells exhibit autoreactivity, enhanced proliferation, increased levels of Th1 and Th2 cytokines, and inhibition of apoptosis. Similar phenomena are observed in the B cells derived from B cell-specific PTEN-deficient (bPtenflox/flox) mice (Suzuki et al., 2001, 2003a). They suggest that PTEN negatively regulates most cellular functions in the immune system. Although PTEN has been considered as a potential therapeutic protein against inflammatory diseases, its inability to enter cells hinders its use for this purpose. Therefore, in an effort to deliver PTEN protein to cells, we investigated the possibility of a protein transduction. In previous studies, we have shown that PEP-1-SOD fusion protein can be efficiently transduced into cells and skin tissue. Moreover, transduced PEP-1-SOD proved enzymatically and biologically active, and efficiently protected against neuronal cell death caused by transient forebrain ischemia (Eum et al., 2004b). Also, we reported that transduced PEP-1-Grb7 fusion protein markedly increased cell viability in macrophage Raw 264.7 cells treated with LPS by inhibition of the COX-2 expression level (An et al., 2007). Recently, we showed that transduced PEP-1-PLP phosphatase (PLPP) fusion protein significantly decreased PLP concentration in PC12 cells pretreated with the vitamin B6 precursors (Lee et al., 2008).
The PEP-1-PTEN fusion protein was expressed, purified and it was found to be nearly homogeneous and greater than 95% pure, as determined by a SDS-PAGE analysis. The expressed and purified PEP-1-PTEN fusion proteins were confirmed by Western blot using an anti-rabbit polyhistidine antibody. Purified PEP-1-PTEN fusion proteins were efficiently transduced into Raw 264.7 cells in a time- and dose-dependent manner. The intracellular stability of transduced PEP-1-PTEN persisted in the cells for 24 h. Although the mechanism of transduction is unclear, protein transduction domain (PTD) fusion protein transduction is a major development in protein therapeutics. Morris et al. (2001) showed that PEP-1 peptide/GFP (green fluorescent protein, 30 kDa) or β-Gal (β-galactosidase, 119 kDa) mixtures transduce into a human fibroblast cell line (HS-68) and into Cos-7 cells by incubating with a PEP-1 peptide carrier and proteins (GFP or β-gal) for 30 min at 37℃. These differences in the time courses of transduction may depend on whether the target protein is fused with the PEP-1 vector or mixed with the PEP-1 peptide. As a result of fusion with the PEP-1 vector, the conformation, polarity, and the molecular shape of a target protein might be altered, which would improve the transduction of fusion proteins into cells.
It is well known that COX-2 and NO produced in macrophages play critical roles in inflammatory diseases (Romanovsky et al., 2006; Lee et al., 2007; Kim et al., 2007). Thus, the inhibition of COX-2 and NO expressions may constitute an effective new therapeutic strategy for the treatment of inflammation and the prevention of inflammatory diseases. To determine whether transduced PEP-1-PTEN can play a biological role in cells, we tested the effects of transduced PEP-1-PTEN on COX-2 expression levels and NO production under LPS exposure. COX-2 expression is induced by a number of stimuli, including cytokines, during the inflammatory response (Carey et al., 2003). Nonsteroidal anti-inflammatory drugs inhibit COX, leading to a marked decrease in prostaglandin synthesis and inflammation (Simon, 1996). The transduced PEP-1-PTEN fusion protein significantly suppressed LPS-induced COX-2 expression levels and NO production in Raw 264.7 cells in a dose-dependent manner.
NF-κB is a transcription factor that controls a number of genes, such as, iNOS and COX-2, TNF-α, and IL-6, which are important for immunity and inflammation (Barnes et al., 1997; Yun et al., 2008). Upon stimulation with LPS, NF-κB is translocated in the cytoplasm as an inactive complex bound to IκB-α, which is phosphorylated and subsequently degraded, and then dissociates to produce activated NF-κB. Therefore, we examined the effect of transduced PEP-1-PTEN on the LPS-induced activation of NF-κB. We found that the translocation of NF-κB was inhibited by transduced PEP-1-PTEN fusion protein in a dose-dependent manner, as well as the phosphorylation and degradation of IκB-α in the cells. In addition, we performed experiments to explore the effect of PEP-1-PTEN fusion protein on the LPS-induced expression levels of iNOS and COX-2 mRNA. The transduced PEP-1-PTEN fusion protein markedly inhibited iNOS and COX-2 mRNA expression levels. Thus, inhibition of these mediators may constitute an effective therapeutic strategy for the prevention of inflammatory reactions and diseases.
Suzuki et al. (2003b) indicates that k5Ptenflox/flox mice, which a keratinocyte-specific null mutation of Pten, exhibit noticeably wrinkled skin, and more than 90% of the mice died within 3 weeks of birth. Skin is continuously exposed to many hazardous environmental agents. PTEN is an essential regulator of normal development and oncogenesis in the skin. Thus, the cell permeable PTEN fusion proteins used in this study may have therapeutic potential against skin inflammation disorders when applied topically. PEP-1 peptide carriers present several advantages for protein therapy, which include the translocation of native proteins, high stability, a lack of toxicity, and a lack of sensitivity to serum. In particular, no toxicity to PEP-1 peptide was observed in several cell lines at up to 1 mM. Although the exact mechanisms of protein transduction are unclear, transduction of the PTEN fused with PEP-1 vector offers more attractive advantages for protein therapy.
In summary, we demonstrated for the first time that human PTEN fused with PEP-1 peptide (PEP-1-PTEN) can be efficiently transduced into Raw 264.7 cells. In addition, transduced PEP-1-PTEN fusion protein significantly suppressed LPS-induced COX-2 expression, NO production. Moreover, the inhibitory effects of PEP-1-PTEN fusion protein were found to be associated with NF-κB inactivation via the blockade of IκB-α phosphorylation. Although the detailed mechanism needs to be further elucidated, our success in the protein transduction of PEP-1-PTEN may be beneficial in developing topical application against inflammatory skin disorders.
Abbreviations
- COX-2:
-
cyclooxygenase-2
- NO:
-
nitric oxide
- PTD:
-
protein transduction domain
- PTEN:
-
phosphatase and tensin homologue deleted on chromosome 10
References
Adams DO, Hamilton TA . The cell biology of macrophage activation . Annu Rev Immunol 1984 ; 2 : 283 - 318
An JJ, Kim SY, Lee SH, Kim DW, Ryu HJ, Yeo SI, Jang SH, Kwon HJ, Kim TY, Lee SC, Poo H, Cho SW, Lee KS, Park J, Eum WS, Choi SY . Transduced PEP-1-Grb7 fusion protein suppressed LPS-induced COX-2 expression . J Biochem Mol Biol 2007 ; 40 : 189 - 195
Barnes PJ, Karin M . Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases . N Engl J Med 1997 ; 336 : 1066 - 1071
Bousquet J, Jeffery PK, Busse WW, Johnosn M, Vignola AM . Asthma: from bronchoconstriction to airways inflammation and remodeling . Am J Respir Crit Care Med 2000 ; 161 : 1720 - 1745
Bradford MA . A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding . Anal Biochem 1976 ; 72 : 248 - 254
Cantley LC, Neel BG . New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/Akt pathway . Proc Natl Acad Sci USA 1999 ; 96 : 4240 - 4245
Carey MA, Germolec DR, Langenbach R, Zeldin DC . Cyclooxygenase enzymes in allergic inflammation and asthma . Prostaglandins Leukot Essent Fatty Acids 2003 ; 69 : 157 - 162
Choi HS, An JJ, Kim SY, Lee SH, Kim DW, Yoo KY, Won MH, Kang TC, Kwon HJ, Kang JH, Cho SW, Kwon OS, Park J, Eum WS, Choi SY . PEP-1-SOD fusion protein efficiently protects against paraquat-induced dopaminergic neuron damage in a Parkinson disease mouse model . Free Radic Biol Med 2006a ; 41 : 1058 - 1068
Choi HS, Lee SH, Kim SY, An JJ, Hwang SI, Kim DW, Yoo KY, Won MH, Kang TC, Kwon HJ, Kang JH, Cho SW, Kwon OS, Choi JH, Park J, Eum WS, Choi SY . Transduced Tat-α-synuclein protects against oxidative stress in vitro and in vivo . J Biochem Mol Biol 2006b ; 39 : 253 - 262
Choi SH, Kim DW, Kim SY, An JJ, Lee SH, Choi HS, Sohn EJ, Hwang SI, Won MH, Kang TC, Kwon HJ, Kang JH, Cho SW, Park J, Eum WS, Choi SY . Transduced human copper chaperone for Cu,Zn-SOD (PEP-1-CCS) protects against neuronal cell death . Mol Cells 2005 ; 20 : 401 - 408
Dunzendorfer S, Meierhofer C, Wiedermann CJ . Signaling in neuropeptide-induced migration of human eosinophils . J Leukoc Biol 1998 ; 64 : 828 - 834
Eum WS, Choung IS, Kim AY, Lee YJ, Kang JH, Park J, Lee KS, Kwon HY, Choi SY . Transduction efficacy of Tatsuperoxide dismutase is enhanced by copper ion recovery of the fusion protein . Mol Cells 2002 ; 13 : 334 - 340
Eum WS, Choung IS, Li MZ, Kang JH, Kim DW, Park J, Kwon HY, Choi SY . HIV-1 Tat-mediated protein transduction of Cu,Zn-superoxide dismutase into pancreatic beta cells in vitro and in vivo . Free Radic Biol Med 2004a ; 37 : 339 - 349
Eum WS, Kim DW, Hwang IK, Yoo KY, Kang TC, Jang SH, Choi HS, Choi SH, Kim YH, Kim SY, Kwon HY, Kang JH, Kwon OS, Cho SW, Lee KS, Park J, Won MH, Choi SY . In vivo protein transduction: Biologically active intact PEP-1-superoxide dismutase fusion protein efficiently protects against ischemic insult . Free Radic Biol Med 2004b ; 37 : 1656 - 1669
Frigas E, Gleich GI . The eosinophil and the pathophysiology of asthma . J Allergy Clin Immunol 1986 ; 77 : 527 - 537
Fugihara S, Ward C, Dransfield I, Hay RT, Uings IJ, Hayes B, Farrow SN, Haslett C, Rossi AG . Inhibition of nuclear factor-kappaB activation un-masks the ability of TNF-alpha to induce human eosinophil apoptosis . Eur J Immunol 2002 ; 32 : 457 - 466
Gleich GJ . Mechanisms of eosinophil-associated inflammation . J Allergy Clin Immunol 2000 ; 105 : 651 - 663
Kim DW, Kim CK, Choi SH, Choi HS, Kim SY, An JJ, Lee SR, Lee SH, Kwon OS, Kang TC, Won MH, Cho YJ, Cho SW, Kang JH, Kim TY, Lee KS, Park J, Eum WS, Choi SY . Tat-mediated protein transduction of human brain pyridoxal kinase into PC12 cells . Biochimie 2005 ; 87 : 481 - 487
Kim JB, Han AR, Park EY, Kim JY, Cho W, Lee J, Seo EK, Lee KT . Inhibition of LPS-induced iNOS, COX-2 and cytokines expression by poncirin through the NF-κB inactivation in Raw 264.7 macrophage cells . Biol Pharm Bull 2007 ; 30 : 2345 - 2351
Kim SY, An JJ, Kim DW, Choi SH, Lee SH, Hwang SI, Kwon OS, Kang TC, Won MH, Cho SW, Park J, Eum WS, Lee KS, Choi SY . Tat-mediated protein transduction of human pyridoxine-5-P oxidase into PC12 cells . J Biochem Mol Biol 2006 ; 39 : 76 - 83
Kwak YG, Song CH, Yi HK, Hwang PH, Kim JS, Lee KS, Lee YC . Involvement of PTEN in airway hyperresopnsiveness and inflammation in bronchial asthma . J Clin Invest 2003 ; 111 : 1083 - 1092
Lee TH, Kwak HB, Kim HH, Lee ZH, Chung DK, Baek NI, Kim J . Methanol extracts of stewartia koreana inhibit cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) gene expression by blocking NF-κB transactivation in LPS-activated Raw 264.7 cells . Mol Cells 2007 ; 23 : 398 - 404
Lee YP, Kim DW, Lee MJ, Jeong MS, Kim SY, Lee SH, Jang SH, Park J, Kang TC, Won MH, Cho SW, Kwon OS, Eum WS, Choi SY . Human brain pyridoxal-5'-phosphate phosphatase (PLPP): protein transduction of PEP-1-PLPP into PC12 cells . BMB Rep 2008 ; 41 : 408 - 413
Li J, Yen C, Liaw K, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodger L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R . PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer . Science 1997 ; 275 : 1943 - 1947
Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M, Zinner R, Hung MC, Steck P, Siminovitch K, Mills GB . The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells . Oncogene 1999 ; 18 : 7034 - 7045
Maehama T, Dixon JE . PTEN/MMAC1, dephosphorylatesthe lipid second messenger, phosphatidylinositol 3,4,5-triphosphate . J Biol Chem 1998 ; 273 : 13375 - 13378
Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG . A fluorometric assay for the measurement of nitrite in biological samples . Anal Biochem 1993 ; 214 : 11 - 16
Morris MC, Depollier J, Mery J, Heitz F, Divita G . A peptide carrier for the delivery of biologically active proteins into mammalian cells . Nat Biotechnol 2001 ; 19 : 1173 - 1176
Morrison DC, Ryan JL . Endotoxins and disease mechanisms . Annu Rev Med 1987 ; 38 : 417 - 432
Myers MP, Pass I, Batty IH, van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK . The lipid phosphatase activity of PTEIN is critical for its tumor suppressor function . Proc Natl Acad Sci USA 1998 ; 95 : 13513 - 13518
Palframan RT, Collins PD, Severs NJ, Rothery S, Williams YJ, Rankin SM . Mechanisms of acute eosinophil mobilization from the bone marrow stimulated by interleukin 5: the role of specific adhesion molecules and phosphatidylinositol 3-kinase . J Exp Med 1998 ; 188 : 1621 - 1632
Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zahringer U, Seydel U, Di Padova F . Bacterial endotoxin: molecular relationships and structure to activity and function . FASEB J 1994 ; 8 : 217 - 225
Romanovsky AA, Ivanov AI, Petresen SR . Microsomal prostaglandin E synthase-1, ephrins, and ephrin kinases as suspected therapeutic targets in arthritis . Ann N Y Acad Sci 2006 ; 1069 : 183 - 194
Simon LS . Actions and toxicity of nonsteroidal anti-inflammatory drugs . Curr Opin Rheumatol 1996 ; 8 : 169 - 175
Smith WL, Dewit DL . Prostaglandin endoperoxide H synthase-1 and -2 . Adv Immunol 1990 ; 62 : 167 - 215
Song HY, Ju SM, Lee JA, Kwon HJ, Eum WS, Jang SH, Choi SY, Park J . Suppression of HIV-1 Tat-induced monocyte adhesiveness by a cell-permeable superoxide dismutase in astrocytes . Exp Mol Med 2007 ; 39 : 778 - 786
Steck PA, Pershouse MA, Jasser SA, Yung WKA, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DHF, Tavtigian SV . Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers . Nat Genet 1997 ; 15 : 356 - 362
Suzuki A, Tsukio-Yamaguchi M, Ohteki T, Sasaki T, Kaisho T, Kimura Y, Yoshida R, Wakeham A, Higuchi T, Fukumoto M, Tsubata T, Ohashi PS, Koyasu S, Penninger JM, Nakano T, Mak TW . T cell-specific loss of Pten leads to defects in central and peripheral tolerance . Immunity 2001 ; 14 : 523 - 534
Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, Sasaki T, Mak TW, Nakano T . Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination . J Exp Med 2003a ; 197 : 657 - 667
Suzuki A, Itami S, Ohishi M, Hamada K, Inoue T, Komazawa N, Senoo H, Sasaki T, Takeda J, Manabe M, Mak TW, Nakano T . Keratinocyte-specific Pten deficiency results in epidermal hyperplasia, accelerated hair follicle morphogenesis and tumor formation . Cancer Res 2003b ; 63 : 674 - 681
Tigani B, Hannon JP, Mazzoni L, Fozard JR . Effects of wortmannin on airways inflammation induced by allergen in actively sensitized Brown Norway rats . Eur J Pharmacol 2001 ; 433 : 217 - 223
Tilley SL, Coffman TM, Koller BA . Mixed message: gene: modulation of inflammation and immune responses by prostaglandins and thromboxanes . J Clin Invest 2001 ; 107 : 191 - 195
Vancheri C, Mastruzzo C, Sortino MA, Crimi N . The lung as a privileged site for the beneficial actions of PEG2 . Trends Immunol 2004 ; 25 : 40 - 46
Vazquez F, Sellers WR . The PTEN tumor suppressor protein: an antagonist of phosphoinositide 3-kinase signaling . Biochim Biophys Acta 2000 ; 1470 : M21 - M35
Wadia J, Dowdy SF . Protein transduction technology . Curr Opin Biotechnol 2002 ; 13 : 52 - 56
Wong CK, Zhang JP, Ip WK, Lam CW . Activation of p38 mitogen-activated protein kinase and nuclear factor-kappaB in tumour necrosis factor-induced eotaxin release of human eosinophils . Clin Exp Immunol 2002 ; 128 : 483 - 489
Yamada KM, Araki M . Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis . J Cell Sci 2001 ; 114 : 2375 - 2382
Yun KJ, Kim JY, Kim JB, Lee KW, Jeong SY, Park HJ, Jung HJ, Cho YW, Yun K, Lee KT . Inhibition of LPS-induced NO and PGE2 production by asiatic acid via NF-κB inactivation in Raw 264.7 macrophages: Possible involvement of the IKK and MAPK pathways . Int Immunopharmacol 2008 ; 8 : 431 - 441
Zhang SY, Park KW, Oh S, Cho HJ, Cho HJ, Park JS, Cho YS, Koo BK, Chae IH, Choi DJ, Kim HS, Lee MM . NF-kappaB decoy potentiates the effects of radiation on vascular smooth muscle cell by enhancing apoptosis . Exp Mol Med 2005 ; 37 : 18 - 26
Acknowledgements
This work was supported by the Next Generation Growth Engine Program Grant (F104AC010002-06A0301-00210) from the Korean Science and Engineering Foundation and by a Regional Innovation Center (RIC) Grant from the Ministry of Commerce and Energy and in part by the Basic Research Promotion Grant (KRF-2005-070-C00091) from Korea Research Foundation.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lee, S., Lee, Y., Kim, S. et al. Inhibition of LPS-induced cyclooxygenase 2 and nitric oxide production by transduced PEP-1-PTEN fusion protein in Raw 264.7 macrophage cells. Exp Mol Med 40, 629–638 (2008). https://doi.org/10.3858/emm.2008.40.6.629
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2008.40.6.629
