
Abstract
For cancer gene therapy, cancer-specific over-expression of a therapeutic gene is required to reduce side effects derived from expression of the gene in normal cells. To develop such an expression vector, we searched for genes over-expressed and/or specifically expressed in cancer cells using bioinformatics and have selected genes coding for protein regulator of cytokinesis 1 (PRC1) and ribonuclease reductase 2 (RRM2) as candidates. Their cancer-specific expressions were confirmed in both breast cancer cell lines and patient tissues. We compared each promoter's cancer-specific activity in the breast normal and cancer cell lines using the luciferase gene as a reporter and confirmed cancer-specific expression of both PRC1 and RRM2 promoters. To test activities of these promoters in viral vectors, the promoters were also cloned into an adeno-associated viral (AAV) vector containing green fluorescence protein (GFP) as the reporter. The GFP expression levels by these promoters were various depending on cell lines tested and, in MDA-MB-231 cells, GFP activities derived from the PRC1 and RRM2 promoters were as strong as that from the cytomegalovirus (CMV) promoter. Our result showed that a vector containing the PRC1 or RRM2 promoter could be used for breast cancer specific overexpression in gene therapy.
Similar content being viewed by others

Introduction
Gene therapy is an attractive treatment to correct defective genes that cause genetic diseases or to kill cancer cells specifically. From more than two decades of research, it is obvious that a successful gene therapy requires several critical components (Lo et al., 2005). For example, a cancer gene therapy regime needs a good therapeutic gene that can specifically kill cancer cells, arrest angiogenesis, inhibit tumor growth or activate cellular immune potential. It also requires a safe and efficient system that can deliver a therapeutic gene into the specific cancer cells and a promoter that exhibits both cancer-specific and strong expression. For target genes, proapoptotic genes such as bcl-2 (Lin et al., 2001), bcl-XS (Ealovega et al., 1996) or death receptor/ligand genes such as TNF-related apoptosis-inducing ligands (Gu et al., 2002) were investigated for cancer gene therapy. For a safe and efficient gene delivery vehicle, various viral and non-viral systems were developed (El-Aneed, 2004; Dass and Choong, 2006; Dobson, 2006). The cytomegalovirus (CMV) promoter has been used since it is one of the strongest promoters in mammalian cells. However, the CMV promoter is not able to differentiate normal and cancer cells for its target gene expression. To utilize toxic or killing proteins in cancer gene therapy, a cancer-specific delivery and/or expression of their genes is critical for safety. Therefore, a better promoter that can exclusively express the target gene in the target cells or at least at much higher level in cancer cells than in normal cells is required for the safe cancer gene therapy.
In order to achieve this goal, several genes have been identified as cancer-specific genes and studied to assess whether or not their promoters could be utilized for development of a cancer-enhanced or -specific gene expression vector. The human telomerase reverse transcriptase (hTERT) promoter was one of the most extensively studied promoters for cancer gene therapy (Gu et al., 2000; Koga et al., 2001; Gu and Fang, 2003) since approximately 90% of human cancer cells are hTERT positive whereas most normal cells except germ cells are hTERT negative (Kim et al., 1994). Baculoviral IAP repeat-containing 5 (BIRC5 or survivin) has been also reported to be overexpressed in common cancers, but not in normal adult tissues (Ambrosini et al., 1997) and investigated for its potential for cancer gene therapy (Chen et al., 2004).
In order to identify other cancer specific genes, we searched published records and databases and chose two genes; protein regulator of cytokinesis 1 (PRC1) and ribonucleotide reductase subunit 2 (RRM2). PRC1 is a mitotic spindle-associated Cdk substrate protein and essential for cell cleavage (Mollinari et al., 2002). PRC1 is negatively regulated by p53 and overexpressed in p53 defective cells (Li et al., 2004), suggesting that the gene is tightly regulated in a cancer-specific manner. RRM2 is a small subunit of ribonucleotide reductase, an enzyme converting ribonucleotides to the corresponding deoxyribonucleotides (Engstrom et al., 1985). RRM2 is overexpressed in breast carcinoma tissue (Jensen et al., 1994).
In order to find promoters suitable for development of a vector for breast cancer gene therapy, we tested activity and specificity of both PRC1 and RRM2 promoters in breast cancer cell lines delivered by as either naked plasmids or adeno-associated virus (AAV)-mediated forms. We employed AAV-mediated system as a gene delivery vector since it has been considered as highly efficient and safe vectors among many developed viral gene delivery systems (Li et al., 2005). Recently, AAV that has been mainly used for treatment of monogenic diseases began to be developed for cancer gene therapy since some AAV serotypes showed cancer tropism and highly safe (Lee et al., 2007; Park et al., 2008).
Here, we report that both PRC1 and RRM2 promoters could be used to develop cancer-specific promoters for breast cancer gene therapy.
Materials and Methods
Cloning of promoters and vector constructions
Promoter regions of PRC1 and RRM2 were amplified by PCR with human MCF7 cell genomic DNA as template. In addition, the BIRC5 promoter was also amplified for a positive control (Table 1). The corresponding PRC1 and BIRC5 PCR products were digested with PstI and NcoI and cloned into pmGL3-B, a modified pGL3-Basic vector (Promega, WI). For RRM2, internal ApaI sites were used instead of PstI. For hTERT, EcoRI sites were added to each end for cloning. We also cloned human TERT promoter (a region spanning from 1695 to 3356 bases of accession number AB016767) into the same reporter plasmid. The resulting reporter plasmids were named pmGL3-PRC1, -RRM2, -TERT and -BIRC5. The pmGL3-B plasmid was constructed by addition of XhoI, BamHI, SalI, BglII, MluI, ApaI, PstI and EcoRI sites between KpnI and HindIII sites in the polycloning sites and deletion of the original BamHI site in the pGL3-Basic. To produce recombinant adeno-associated viral vectors (rAAV) containing CMV or a cancer-specific promoter, we made pAAV plasmid constructs using pmGL3-PRC1, -RRM2 and -BIRC5. The pAAV-CMV-GFP was prepared from pAAV-FIX as previously described (Cho et al., 2007). The pAAV plasmid constructs containing three different cancer-specific promoters were subcloned by replacing CMV of pAAV-CMV-GFP with the corresponding SalI and NcoI fragment of PRC1, RRM2 or BIRC5 derived from pmGL3-PRC1, -RRM2 or -BIRC5, respectively, to make pAAV-PRC1-GFP, pAAV-RRM2-GFP or pAAV-BIRC5-GFP.
Cell culture, transfection and luciferase assay
MCF7, T47D and MDA-MB-231 human breast cancer cell lines were cultured in DMEM medium containing 10% FBS. MCF10A, normal human breast cells, were cultured in DMEM/F12 medium supplemented with 20 ng/ml EGF, 500 ng/ml hydrocortisone, 100 ng/ml of cholera toxin, 10 ng/ml insulin, 2mM L-glutamine and 5% FBS (Yang et al., 2004). All cell lines were incubated in 5% CO2 at 37℃.
For transient transfection, cells were seeded in wells of 24 well plates at 5 × 104, 0.7 × 104 or 1.5 × 105 cells/well for MDA-MB-231, T47D or MCF10A cells, respectively. MCF7 cells were seeded at 7 × 104 cells/well in 48 well plates a day before transfection. For 24 well plates, 350 ng of each of pmGL3 reporter and 50 ng of pRL-TK plasmids were used. For 48 well plates, plasmid DNA amount was reduced by half. Cells were harvested and assayed for reporter activity 2 days after transfection. Transient transfections and luciferase assay were carried out with LipofectAMINE2000 (Invitrogen, CA) and with Dual-Luciferase Reporter Assay System (Promega), respectively, as recommended by the manufacturers. pRL-TK was included to normalize transfection efficiency for all cell lines except MCF10A cells.
RT-PCR and western blot analysis
For RT-PCR, total RNAs were isolated from corresponding cells and RT-PCR was carried out as described (Jeong et al., 2006) using specific primer sets (PRC1: forward 5' CTATGATATTGACAGTGCCTCAGTGC and reverse 5' TATTTGCAACCTGTCCCAGAGCTCTCG; RRM2: forward 5' GCTTGGTCGACAAGGAGAAC and reverse 5' CCTCTGCCTTCTTATACATCTG). The PCR was carried out at 94℃ for 5 min, 35 cycles of 94℃ for 30 s, 65℃ for 30 s and 72℃ for 30 s followed by 72℃ for 2 min for the PRC1 gene. For the RRM2 gene, the annealing temperature and amplification cycle were changed to 55℃ and 40 cycles, respectively. For a quantitative control, a GAPDH specific primer set (forward: 5' GCTGAGTACGTCGTGGAGTC and reverse: 5' AGGCATTGCTGATGATCTTG) was included for each PCR reaction. The cDNA amount in each PCR reaction used as template was adjusted so that level of each GAPDH used as an internal control was similar to among cell lines. For western analysis, cells were lysed by a Laemmli buffer, the lysates were subjected to SDS-PAGE and specific proteins were detected using anti-PRC-1 (Biolegend, CA, No.629001) or anti-RRM2 (Abnova, Taiwan, H00006241-M01) antibodies. Anti-β-actin (Sigma Co. A5441) was used for a quantitative control.
Immunohistochemistry
The materials were 85 cases of breast carcinomas embedded in 6 tissue microarray blocks obtained from paraffin blocks of known breast carcinoma cases during the period of Jan. 1999 to Dec. 2001. In 25 of 85 cases, normal-looking breast tissues were observed next to the cancer tissue. Tissue sections were deparaffinized with xylene and rehydrated in decreasing concentrations of alcohols. After rinsing with water, antigen retrieval was performed using target retrieval solution (pH 9.0) at 95-99℃ for 30 min. The following steps were performed in the Autoimmuostainers (DAKO, Carpinteria, CA) using Envision Kit (DAKO). Endogenous peroxidase activity was blocked by incubating slides with 5% hydrogen peroxide in PBS for 10 min. Sections were incubated with specific primary PRC1 and RRM2 antibodies for 1 h, followed by rinsing. The slides were incubated with secondary antibody and then treated with DAB substrate for 3-5 min. After rinsing, sections were counterstained with Mayer's hematoxylin for 10 s. Finally, slides were dehydrated, coverslipped with Crystal mount medium and observed to visualize expression of PRC1 and RRM2 proteins.
Recombinant AAV (rAAV) vector production, transduction and FACS analysis
HEK293 or 293T cells were maintained as described previously (Nenoi et al., 2006) and transiently co-transfected with the following three plasmids using the calcium phosphate precipitation methods: (i) a pAAV-GFP plasmid containing GFP cDNA under CMV or tumor specific promoter, (ii) an adenoviral helper plasmid (pHelper plasmid, Stratagene, La Jolla, CA), (iii) an AAV helper plasmid encoding AAV serotype 2 (AAV2) rep/cap genes (pAAV-RC, Stratagene). The rAAV serotype 2 (rAAV2) vectors such as rAAV2-CMV-GFP, rAAV2-PRC1-GFP, rAAV2-RRM2-GFP and rAAV2-BIRC5-GFP were purified by repeated CsCl gradient centrifugation and produced in excess of the titer of 105 viral genomes per cell as determined by a real time PCR (Mini Opticon, CFD3120, Bio-Rad, CA) and ELISA (AAV2 Titration ELISA System, PROGEN, Heidelberg, Germany).
To evaluate cancer-specific promoter activities, MCF7, MDA-MB-231 and T47D human breast cancer cells along with MCF10A normal cells were seeded at 1 × 104 cells/well in 12-well plates and infected with 200,000 multiplicity of infection (MOI) of rAAV vectors for 72 h. GFP expressions in the rAAV-transduced cells were monitored by a fluorescent microscopy (CKX41, OLYMPUS, Tokyo, Japan) and also measured by a FACS flow cytometry equipped with a 488 nm argon laser (FACS Calibur, Becton Dickinson, CA). For a FACS flow cytometry, the infected cells were trypsinized, washed with ice-cold PBS and fixed with 4% paraformaldehyde. The gate was set to exclude cell debris and death cells. Data from 10,000 cells/sample were collected and the results were shown in a histogram of fluorescence intensity (X-axis) and cell counts (Y-axis). The fluorescence intensity of the negative control cells of mock infection was used to distinguish between the infected population and the uninfected population by setting M1 for each cell line, since the infected population of the cells emitted a fluorescent signal ranging M1. To verify that differences of GFP expression in the rAAV2-transduced cells were attributed to those of promoter activity, AAV transduction efficiencies of each tested cell of MCF7, T47D, MDA-MB-231 and MCF10A cell lines were determined as follows. The viral genomes in the infected cells with rAAV2-GFP vectors containing CMV, BIRC5, RRM2 or PRC1 promoter were isolated by the Hirt method (Eizuru et al., 1984) and the infected AAV genomes were quantified using semi-quantitative PCR amplifications.
Results
PRC1 and RRM2 are overexpressed in breast cancer cells
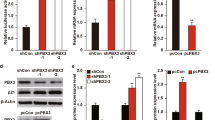
The expression levels of both PRC1 and RRM2 were determined in normal breast cell line (MCF10A) and a pair of breast cancer cell lines (MCF7 and T47D) at mRNA and protein levels (Figure 1). To adjust the same amount of RNA or proteins used for analysis, GAPDH or β-actin was employed as an internal quantitative control, respectively. At mRNA level, both PRC1 and RRM2 showed approximately 1.5- and 2-fold activation, respectively (Figure 1A). The expression levels of each protein of cancer cell lines were also increased remarkably compared to those of normal cells (Figure 1B). MCF10A cells exhibited almost no protein expression of RRM2 or PRC1, whereas MCF7 and T47D showed strong protein expression.

Expression level of PRC1 and RRM2 at both mRNA (A) and protein (B) levels. (A) Total RNAs were isolated from the indicated breast normal (MCF10A) and cancer (MCF7 and T47D) cell lines and used for reverse transcription. PCR was carried out using a specific PRC1 or RRM2 primer set. For quantitative comparison, a specific GAPDH primer set was mixed with the gene specific primer set for each PCR reaction. (B) Lysates of indicated cell lines were subjected to SDS-PAGE following western blot analysis using specific antibody. For a quantitative control, anti-β-actin antibody has been used.
To further test expression levels of both genes in cancer vs normal breast tissues, immunohistochemistry study was carried out using tissues in paraffin blocks prepared from human breast carcinoma cases. We investigated 85 breast cancer tissue samples for PRC1 and RRM2 expression. Both PRC1 and RRM2 expressions were detected in almost half of cancer samples, 41 (48%) or 49 (58%) out of 85 samples, respectively. However, neither RRM2 nor PRC1 was detected in normal cells present next to cancer cells. The representative expression patterns in cancer and normal tissues were shown in Figure 2. In these 85 samples, estrogen receptor α, an important marker of breast cancer, was detected in approximately 50% of the cancer tissues tested. Therefore, it could be concluded that both RRM2 and PRC1 were specifically overexpressed in breast cancer tissue.

Representative immunohistochemistry results of PRC1 and RRM2 expression in human breast carcinoma samples. The frozen human breast cancer (B, D) and normal tissue samples (A, C) were stained with PRC1 (A, B) or RRM2 (C, D) antibodies. The magnification is 100-fold and nuclei were stained blue.
PRC1 and RRM2 show cancer-specific promoter activity in breast cancer cell lines
To determine whether promoters of PRC1 and RRM2 could be utilized for cancer-specific expression, we cloned the promoter region of each gene into modified pGL3-Basic and named the resulting plasmid pmGL3-PRC1 or -RRM2. Each of these plasmids were transiently transfected into breast normal and cancer cell lines and their luciferase activities were compared to one expressed from the cancer-specific promoter such as BIRC5 or hTERT (Ambrosini et al., 1997; Gu and Fang 2003; Chen et al., 2004). In normal MCF10A cell lines, both PRC1 and RRM2 as well as hTERT and BIRC5 promoters exhibited no activation of the luciferase whereas the CMV promoter exhibited approximately 18-fold activation (Figure 3A). In MCF7 and T47D breast cancer cell lines, both PRC1 and RRM2 promoters increased the luciferase reporter activity approximately 4- to 10-fold (Figure 3B and D). Interestingly, in MDA-MB-231 cell line, both PRC1 and RRM2 exhibited remarkable activation of the reporter, much better than that of the BIRC5 (Figure 3C). In addition, human TERT promoter activity was quite low compared to those of PRC1 and RRM2 in MCF7 and MDA-MB-231 cancer cell lines (Figure 3B and C).

Cancer-specific activation of the reporter gene by both PRC1 and RRM2 promoters. pmGL3-PRC1, -RRM2, -BIRC5, TERT or empty pmGL3 (pGL3-basic) vector was transiently transfected into normal (A: MCF10A) or cancer (B: MCF7; C: MDA-MB-231 or D: T47D) breast cell lines. pRL-TK was also co-transfected to normalize transfection efficiency except MCF10A. For MCF10A cells, only reporter plasmids were transfected and the average of three separate experiments with triplicates was shown. Forty-eight hours after transfection, cells were harvested and their luciferase activity was measured. The transfection experiments were carried out more than twice in triplicate and one representative result was shown. The CMV promoter was included in MCF10A cell line to confirm its strong activity in the normal cell line. In T47D cell line, TERT was omitted.
Cancer-specific expression in the transduced cells with rAAV2 containing PRC1 or RRM2 promoter
To further determine cancer-specific expression of the PRC1 or RRM2 promoter, we examined their activities in AAV-mediated gene delivery system that has been reported as a safe and highly efficient vector for cancer gene therapy (Lee et al., 2007; Park et al., 2008). The rAAV2-PRC1-GFP, rAAV2-RRM2-GFP and rAAV2-BIRC5-GFP were infected into each of MCF7, T47D and MDA-MB-231 cancer cell lines along with the positive control rAAV2 containing the CMV promoter, rAAV2-CMVGFP. These rAAV2 vectors were also transduced into MCF10A normal cells. To evaluate promoter activities and to determine cancer-specific transcriptional targeting, GFP expressions in rAAV2-infected cells were measured using an FACS flow cytometry. The GFP expression derived from rAAV2-CMV-GFP was assumed to be 100, and relative GFP expressions derived from other vectors were plotted in the bar graphs (Figure 4B). The experiment was repeated three times in triplicate and the results plotted in Figure 4B were the average values of independently conducted experiments. One of the representative FACS data in a histogram was also presented in Figure 4A. To determine the AAV transduction efficiencies in each case, amount of AAV genome in the transduced cells was measured by a semi-quantitative PCR. The result indicated that the AAV transduction efficiencies in the cells infected with different types of rAAV2 vectors were quite similar among them, suggesting that the differences of GFP observed were resulted from those of promoters' activities (data not shown). In the transduced normal MCF10A cells, the PRC1, RRM2 and BIRC5 showed low activation of the GFP reporter, approximately 10-30% of that of the CMV promoter. When these rAAV2 vectors were transduced to the breast cancer cell lines, they exhibited broad range of expression activity. In MDA-MB-231 cells, both PRC1 and RRM2 promoters showed activity comparable to that of the CMV promoter. However, these promoters showed significantly weaker activity in T47D cells and moderate effect in MCF7 cells. We also tested the human TERT and the synthetic human TERT (E3-H-T) (Kim et al., 2006) in our AAV-mediated system and observed that both promoters showed very weak activation compared to both PRC1 and RRM2 promoters in HeLa cells (Park et al., 2007). The overall relative activity in our AAV system was in the order of CMV, BIRC5, PRC1 and RRM2 promoters.

The GFP expression in rAAV2-infected breast normal and cancer cell lines. The promoter activity of the rAAV2 vector containing the GFP reporter gene under cancer-specific promoters of PRC1, RRM2 or BIRC5 was compared to that under the CMV promoter in breast normal (MCF10A) and cancer (MCF7, T47D and MDA-MB-231) cell lines. The cells were seeded at 1 × 104 cells/well in 12-well plates and infected with rAAV2 vectors of 2 × 105 MOI for 72 h. The GFP expressions in the transduced cells were measured using a FACS flow cytometry and the experiment was repeated three times in triplicate. (A) One of representative FACS data collected from 10,000 cells/sample was presented in a histogram of fluorescence intensity (X-axis) and cell counts (Y-axis). The fluorescence intensity of the (-) control cell of mock infection was used to distinguish the rAAV-infected population from the uninfected population by setting M1 for each tested cell line, since the rAAV-infected cells emitted a fluorescent signal ranging M1. (B) The relative GFP expression. The GFP expression in each case was normalized by transduction efficiency of each rAAV measured by semi-quantitative PCR of AAV genomic DNA. The GFP expression derived from rAAV2-CMV-GFP was considered to be 100, and relative GFP expressions in the infected cells with rAAV2 containing the PRC1, RRM2 or BIRC5 promoter were plotted in the bar graph. The average values of the independently repeated experiments were applied and plotted in the graph.
Discussion
In this study, we tested whether PRC1 or RRM2 promoter could be employed for the development of breast cancer-enhanced gene expression. Both PRC1 and RRM2 proteins were highly expressed in the breast cancer cell lines, MCF7 and T47D, compared to their expressions in MCF10A normal cells. Our result suggested that both PRC1 and RRM2 are regulated at the transcription as well as translation level since differences of both genes between normal and cancer cell lines are greater at the protein level than the mRNA level (Figure 1). Actually, expression of RRM2 has been already reported to be regulated at the translation level by presence of four AUG codons at upstream of the translation initiation site (Dong et al., 2005). The overexpressions of PRC1 and RRM2 were confirmed by immunohistochemistry assay using 85 human breast cancer tissue samples. Furthermore, both promoters exhibited considerable cancer-specific activity when they were delivered to cells either as naked plasmid or packed in the AAV system. Especially, the cancer specific activity of the PRC1 promoter in rAAV2 vectors was comparable to the strong CMV promoter in MDA-MB-231 or MCF-7 breast cancer cell lines (Figure 4). The exact reason why the PRC1 promoter activity in rAAV vectors showed stronger activity compared to the naked plasmid is not clear at this point. It may be due to the difference of the assay methods we applied for both experiments such as the delivery system or the reporter genes. However, it is unlikely that a part of AAV genome in rAAV2 vectors may affect promoter activities since four types of rAAV2 vectors are only composed of AAV2 inverted terminal repeats (145-bp) and the essential mammalian expression cassette containing GFP cDNA. Interestingly, all of tested promoters showed remarkable strong activity in the MDA-MB-231 cell line (Figures 3C and 4B). Since the MDA-MB-231 cell line is highly metastatic in contrast to two other cell lines, MCF-7 and T47D, which are poorly metastatic (Sharma et al., 2006), it can be speculated that the cell line specific differences of these promoters' activities might be related to their capabilities for metastasis. Otherwise, it might be due to the status difference of important regulators such as p53 or estrogen receptor (ER) α among the cell lines. For example, p53 has been reported to down-regulate PRC1 expression (Li et al., 2004) and known to be mutated in the MDA-MB-231, but not in the MCF7 cell line (Vojtesek and Lane, 1993; Su et al., 1998). Similarly, MCF7 and MDA-MB-231 were reported as positive and negative, respectively, for ERα which is a strong transcription factor expressed in breast cancer (Chien et al., 1999).
Previously, several cancer-specific promoters including the TERT (Gu et al., 2000; Gu and Fang, 2003), the synthetic hTERT promoter (Kim et al., 2006), tandem hypoxia-responsive elements (HRE) with the enhancerless SV40 promoter (Ruan et al., 2001), the alpha-fetoprotein promoter (Su et al., 1996), calcitonin gene regulatory sequence (Jiang et al., 2001) and the glucose transporter isoform 1 promoter (Sieger et al., 2004) have been identified and examined for cancer-specific expression. We tested the TERT promoter as naked plasmids in MCF7 and MDA-MB-231 cells, but the result showed weaker activation than those of both PRC1 and RRM2 promoters in the same transfection condition (Figure 3). We also examined activities of human TERT or the synthetic human TERT promoters in the AAV-mediated gene delivery system. They all showed certain degree of tumor-enhanced expression in HeLa cells; however, their relative promoter activities were very low and could reach that of neither CMV nor one of PRC1 and RRM2 promoters (data not shown).
Taken together, our results conclusively suggest that both promoters of PRC1 and RRM2 could be used to develop a transcriptional targeting vector of breast cancer gene therapy.
Abbreviations
- AAV:
-
adeno-associated viral (virus)
- BIRC5:
-
Baculoviral IAP repeat-containing 5
- CMV:
-
cytomegalovirus
- GFP:
-
green fluorescence protein
- PRC1:
-
protein regulator of cytokinesis 1
- RRM2:
-
ribonuclease reductase 2
- TERT:
-
telomerase reverse transcriptase
References
Ambrosini G, Adida C, Altieri DC . A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma . Nat Med 1997 ; 3 : 917 - 921
Chen JS, Liu JC, Shen L, Rau KM, Kuo HP, Li YM, Shi D, Lee YC, Chang KJ, Hung MC . Cancer-specific activation of the survivin promoter and its potential use in gene therapy . Cancer Gene Ther 2004 ; 11 : 740 - 747
Chien PY, Ito M, Park Y, Tagami T, Gehm BD, Jameson JL . A fusion protein of the estrogen receptor (ER) and nuclear receptor corepressor (NCoR) strongly inhibits estrogen-dependent responses in breast cancer cells . Mol Endocrinol 1999 ; 13 : 2122 - 2136
Cho YH, Park H, Cho ES, Kim WJ, Kang BS, Park BY, Kim YJ, Lee YI, Chang SI, Park K . A novel way of therapeutic angiogenesis using an adeno-associated virus-mediated angiogenin gene transfer . Exp Mol Med 2007 ; 39 : 412 - 418
Dass CR, Choong PF . Selective gene delivery for cancer therapy using cationic liposomes: in vivo proof of applicability . J Control Release 2006 ; 113 : 155 - 163
Dobson J . Gene therapy progress and prospects: magnetic nanoparticle-based gene delivery . Gene Ther 2006 ; 13 : 283 - 287
Dong Z, Liu Y, Zhang JT . Regulation of ribonucleotide reductase M2 expression by the upstream AUGs . Nucleic Acids Res 2005 ; 33 : 2715 - 2725
Ealovega MW, McGinnis PK, Sumantran VN, Clarke MF, Wicha MS . bcl-xs gene therapy induces apoptosis of human mammary tumors in nude mice . Cancer Res 1996 ; 56 : 1965 - 1969
Eizuru Y, Inagawa S, Minamishima Y . Application of "Hirt supernatant" DNA to the molecular epidemiology of cytomegalovirus infections . J Clin Microbiol 1984 ; 20 : 1012 - 1014
El-Aneed A . An overview of current delivery systems in cancer gene therapy . J Control Release 2004 ; 94 : 1 - 14
Engstrom Y, Eriksson S, Jildevik I, Skog S, Thelander L, Tribukait B . Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits . J Biol Chem 1985 ; 260 : 9114 - 9116
Gu J, Kagawa S, Takakura M, Kyo S, Inoue M, Roth JA, Fang B . Tumor-specific transgene expression from the human telomerase reverse transcriptase promoter enables targeting of the therapeutic effects of the Bax gene to cancers . Cancer Res 2000 ; 60 : 5359 - 5364
Gu J, Zhang L, Huang X, Lin T, Yin M, Xu K, Ji L, Roth JA, Fang B . A novel single tetracycline-regulative adenoviral vector for tumor-specific Bax gene expression and cell killing in vitro and in vivo . Oncogene 2002 ; 21 : 4757 - 4764
Gu J, Fang B . Telomerase promoter-driven cancer gene therapy . Cancer Biol Ther 2003 ; 2 : S64 - S70
Jensen RA, Page DL, Holt JT . Identification of genes expressed in premalignant breast disease by microscopy-directed cloning . Proc Natl Acad Sci USA 1994 ; 91 : 9257 - 9261
Jeong H, Kim MS, Kim SW, Kim KS, Seol W . Regulation of tyrosine hydroxylase gene expression by retinoic acid receptor . J Neurochem 2006 ; 98 : 386 - 394
Jiang S, Altmann A, Grimm D, Kleinschmidt JA, Schilling T, Germann C, Haberkorn U . Tissue-specific gene expression in medullary thyroid carcinoma cells employing calcitonin regulatory elements and AAV vectors . Cancer Gene Ther 2001 ; 8 : 469 - 472
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW . Specific association of human telomerase activity with immortal cells and cancer . Science 1994 ; 266 : 2011 - 2015
Kim SJ, Lee HS, Shin JH, Kim CG, Jeong S, Park K, Choe H, Lee H . Preferentially enhanced gene expression from a synthetic human telomerase reverse transcriptase promoter in human cancer cells . Oncol Rep 2006 ; 16 : 975 - 979
Koga S, Hirohata S, Kondo Y, Komata T, Takakura M, Inoue M, Kyo S, Kondo S . FADD gene therapy using the human telomerase catalytic subunit (hTERT) gene promoter to restrict induction of apoptosis to tumors in vitro and in vivo . Anticancer Res 2001 ; 21 : 1937 - 1943
Lee HS, Shin OK, Kim SJ, Lee WI, Jeong S, Park K, Choe H, Lee H . Efficient gene expression by self-complementary adeno-associated virus serotype 2 and 5 in various human cancer cells . Oncol Rep 2007 ; 18 : 611 - 616
Li C, Lin M, Liu J . Identification of PRC1 as the p53 target gene uncovers a novel function of p53 in the regulation of cytokinesis . Oncogene 2004 ; 23 : 9336 - 9347
Li C, Bowles DE, van Dyke TSamulski RJ . Adeno-associated virus vectors: potential applications for cancer gene therapy . Cancer Gene Ther 2005 ; 12 : 913 - 925
Lin J, Page C, Jin X, Sethi AO, Patel R, Nunez G . Suppression activity of pro-apoptotic gene products in cancer cells, a potential application for cancer gene therapy . Anticancer Res 2001 ; 21 : 831 - 839
Lo HW, Day CP, Hung MC . Cancer-specific gene therapy . Adv Genet 2005 ; 54 : 235 - 255
Mollinari C, Kleman JP, Jiang W, Schoehn G, Hunter T, Margolis RL . PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone . J Cell Biol 2002 ; 157 : 1175 - 1186
Nenoi M, Daino K, Ichimura S, Takahash S, Akuta T . Low-dose radiation response of the p21WAF1/CIP1 gene promoter transduced by adeno-associated virus vector . Exp Mol Med 2006 ; 38 : 553 - 564
Park K, Cho YH, Kim WJ, Seol W, Cho ES, Kim YJ, Cho SH, Kim HJ, Kang BS, Ji WJ . Targeted cancer gene therapy using AAV2-mediated anti-angiogenesis approaches . Molecular Therapy 2007 ; 15 : S244 -
Park K, Kim WJ, Cho YH, Lee YI, Lee H, Jeong S, Cho ES, Chang SI, Moon SK, Kang BS, Kim YJ, Cho SH . Cancer gene therapy using adeno-associated virus vectors . Front Biosci 2008 ; 2008 : 2653 - 2659
Ruan H, Su H, Hu L, Lamborn KR, Kan YW, Deen DF . A hypoxia-regulated adeno-associated virus vector for cancer-specific gene therapy . Neoplasia 2001 ; 3 : 255 - 263
Sharma MR, Koltowski L, Ownbey RT, Tuszynski GP, Sharma MC . Angiogenesis-associated protein annexin II in breast cancer: selective expression in invasive breast cancer and contribution to tumor invasion and progression . Exp Mol Pathol 2006 ; 81 : 146 - 156
Sieger S, Jiang S, Kleinschmidt J, Eskerski H, Schonsiegel F, Altmann A, Mier W, Haberkorn U . Tumor-specific gene expression using regulatory elements of the glucose transporter isoform 1 gene . Cancer Gene Ther 2004 ; 11 : 41 - 51
Su H, Chang JC, Xu SM, Kan YW . Selective killing of AFP-positive hepatocellular carcinoma cells by adeno-associated virus transfer of the herpes simplex virus thymidine kinase gene . Hum Gene Ther 1996 ; 7 : 463 - 470
Su ZZ, Madireddi MT, Lin JJ, Young CS, Kitada S, Reed JC, Goldstein NI, Fisher PB . The cancer growth suppressor gene mda-7 selectively induces apoptosis in human breast cancer cells and inhibits tumor growth in nude mice . Proc Natl Acad Sci USA 1998 ; 95 : 14400 - 14405
Vojtesek B, Lane DP . Regulation of p53 protein expression in human breast cancer cell lines . J Cell Sci 1993 ; 105 : 607 - 612
Yang L, Cao Z, Li F, Post DE, Van Meir EG, Zhong H, Wood WC . Tumor-specific gene expression using the survivin promoter is further increased by hypoxia . Gene Ther 2004 ; 11 : 1215 - 1223
Acknowledgements
This study was supported by grants from the Korean Ministry of Science and Technology (M10534040001-05N3404-00110), the Institute for Brain Science and Technology (IBST Grant 2006 and 2007) and the New Faculty Supporting Grant from Inje University to W. S., as well as a grant from the Korean Ministry of Science and Technology (M10534040005-05N3404-00511) to K. P.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yun, H., Cho, YH., Moon, Y. et al. Transcriptional targeting of gene expression in breast cancer by the promoters of protein regulator of cytokinesis 1 and ribonuclease reductase 2. Exp Mol Med 40, 345–353 (2008). https://doi.org/10.3858/emm.2008.40.3.345
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2008.40.3.345
Keywords
This article is cited by
-
Overexpression of protein regulator of cytokinesis 1 facilitates tumor growth and indicates unfavorable prognosis of patients with colon cancer
Cancer Cell International (2020)
-
Regulation of proliferation and cell cycle by protein regulator of cytokinesis 1 in oral squamous cell carcinoma
Cell Death & Disease (2018)
-
Identification and functional characterization of glioma-specific promoters and their application in suicide gene therapy
Journal of Neuro-Oncology (2011)
