
Abstract
Mitochondrial respiratory chain complex I consists of 44 different subunits and can be subgrouped into three functional modules: the Q-, the P- and the N-module. NDUFAF4 (C6ORF66) is an assembly factor of complex I that associates with assembly intermediates of the Q-module. Via exome sequencing, we identified a homozygous missense variant in a complex I-deficient patient with Leigh syndrome. Supercomplex analysis in patient fibroblasts revealed specifically altered stoichiometry. Detailed assembly analysis of complex I, indicative of all of its assembly routes, showed an accumulation of parts of the P- and the N-module but not the Q-module. Lentiviral complementation of patient fibroblasts with wild-type NDUFAF4 rescued complex I deficiency and the assembly defect, confirming the causal role of the variant. Our report on the second family affected by an NDUFAF4 variant further characterizes the phenotypic spectrum and sheds light into the role of NDUFAF4 in mitochondrial complex I biogenesis.
Similar content being viewed by others


Introduction
Mitochondrial respiratory chain complex I (CI, NADH:ubiquinone oxidoreductase) is the largest component of the oxidative phosphorylation (OXPHOS) system. It is L-shaped and can be subgrouped into three functional modules: the Q-module (ubiquinone reduction) and the N-module (NADH dehydrogenase), which form the matrix arm and the P-module (proton pumping), which makes up the membrane-integrated arm.
CI biogenesis is an intricate process: 37 nuclear-encoded and 7 mitochondrial-encoded subunits form numerous assembly intermediates that are assembled in several parallel and sequential steps to build the fully assembled complex. Subsequently, CI is integrated into supercomplexes, in which it associates with either a complex III (CIII)-dimer (I/III2-supercomplexes) or with a CIII-dimer and one or multiple copies of complex IV (CIV, I/III2/IVn-supercomplexes).1
CI assembly depends on the action of several assembly factors that associate with assembly intermediates but are not part of the holoenzyme itself.
NDUFAF4 (C6ORF66) has been categorized as an assembly factor of the Q-module because it associates with Q-module subassemblies during CI biogenesis.1 However, its exact function has not yet been elucidated. So far, one NDUFAF4 variant that affected protein function has been described in a single family.2 Here, we present a patient with a novel NDUFAF4 variant, expand the phenotype associated with NDUFAF4 deficiency and analyze its role in CI biogenesis.
Materials and methods
Results
Case report
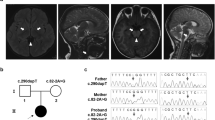
The patient is the first child of consanguineous (first cousins) parents of Pakistani ethnicity. He was born at 38 weeks gestation after induction of labor due to concerns regarding intrauterine growth retardation. He weighed 2406 g.
At 7 months of age, he was presented to the local pediatric services due to a decline in his milestones. He smiled infrequently, was unable to roll over, sit unsupported and did not babble. He was a non-dysmorphic, underweight child with irritability. He had central hypotonia and normal to slightly increased limb tone with easily elicited tendon knee reflexes and no clonus.
Brain MRI revealed diffuse bilateral signal alterations in the basal ganglia and thalami, indicative of Leigh syndrome. An EEG showed generalized slowing with multifocal spikes consistent with an epileptogenic focus. There was no cardiovascular, renal or hepatic disease.
Urine organic acid analysis revealed increased lactate, ketones and Krebs cycle metabolites. Plasma lactate was 10.6 mmol/l and CSF lactate was 6.1 mmol/l (reference range: 0.8–1.8 mmol/l). Patient-derived skin fibroblasts were examined for the activity of the respiratory chain enzymes, which showed isolated CI deficiency (32 mU/U of citrate synthase, reference range: 163–599) with activities of complexes II, III, IV and V within reference range.
Two years after the initial examination, the patient continues to have profound developmental delay, requires feeding through a PEG-tube and has episodic seizures.
Whole-exome sequencing identifies variants in NDUFAF4
Via whole-exome sequencing, a homozygous missense variant in the NDUFAF4 gene (NM_014165.3) was identified: c.7G>C; p.(Ala3Pro). This variant is not present in the ExAC-database (http://exac.broadinstitute.org), the 1000-genomes-database (http://www.internationalgenome.org) or our in-house-database. The novel variant and the previously published variant have been uploaded to 'Leiden Open Variation Database' (http://www.lovd.nl/NDUFAF4): variant IDs #0000170856 and #0000174214; individual IDs #00104960 and #00107834.
Variant prediction softwares predicted different functional effects of this mutation: Mutation taster (http://www.mutationtaster.org/):3 'polymorphism' (probability: 0,84); PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/):4 'probably damaging' (score: 0.991); SIFT (http://sift.jcvi.org/):5 'damaging' (SIFT score 0.04). Sequence alignment showed that the exchanged alanine in position 3 of the amino acid sequence is conserved in mammals (Supplementary Figure 1).
The NDUFAF4 variant leads to isolated CI deficiency that can be rescued via lentiviral complementation with wild-type NDUFAF4
Lentiviral complementation with C-terminally V5-tagged wild-type NDUFAF4 was performed to prove that the NDUFAF4 variant affects protein function (Figure 1). CI enzymatic activity was rescued by complementation with NDUFAF4 but not by mock transfection with V5-tagged GFP (Figure 1a). Complementation also rescued reduced levels of holo-CI, analyzed by blue native polyacrylamide gel electrophoresis (BN-PAGE)/immunoblotting using n-dodecyl-β-D-maltoside lysed mitochondrial protein (Figure 1b). CI subunit levels, analyzed by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), also increased in complemented patient fibroblasts (Figure 1c). NDUFAF4 was not detectable in patient cells.

The NDUFAF4 variant causes isolated complex I deficiency that can be rescued by lentiviral complementation with wild-type NDUFAF4. (a) Complex I enzymatic activity in control (Ctrl) and patient (Pat) cells either transfected with C-terminally V5-tagged wild-type NDUFAF4 (+AF4) or with mock transfected with C-terminally V5-tagged GFP (+GFP). Values are given relative to the activity of citrate synthase (CS) and as average value of two independent experiments. The error bars indicate standard deviation. Lentiviral complementation with wild-type NDUFAF4, but not mock complementation with GFP, increases complex I activity in patient fibroblasts into reference range (163–599 mU/U CS). (b) Non-denaturing BN-PAGE electrophoresis/immunoblotting analysis of mitochondrial protein (lysed with n-dodecyl-β-d-maltoside) from control and patient fibroblasts either transfected with NDUFAF4 or mock transfected with GFP. Holo-complex I (CI, NDUFS3) levels in patient fibroblasts are severely reduced in comparison to control fibroblasts. Complementation with wild-type NDUFAF4 increases holo-complex I levels. The other OXPHOS complexes are not negatively affected: complex V (d subunit, CVdSU), complex III (CIII, UQCRFS1), complex IV (CIV, COX4) and complex II (CII, SDHA). (c) Immunoblotting analysis of total cell lysates after SDS-PAGE with antibodies against subunits representative of the OXPHOS complexes. Levels of complex I subunits representative for each functional module (NDUFV2—N-module, NDUFB11—P-module, NDUFS3—Q-module) are decreased in patient cells and rescued by complementation with wild-type NDUFAF4. Analysis of NDUFAF4, GFP and V5 confirm successful lentiviral complementation. The position of the 25 kDa molecular weight marker is indicated on the right in blots showing the signals of the NDUFAF4 antibody, the V5 antibody and GFP antibody, respectively. Tubulin serves a loading control.
Lack of CI in NDUFAF4-deficient patient fibroblasts causes altered supercomplex stoichiometry
Since CI forms supercomplexes with CIII and CIV, and the levels of the latter two were not changed despite CI deficiency, we analyzed supercomplex stoichiometry. In order to maintain supercomplex interactions, mitochondrial protein was solubilised with the mild detergent digitonin rather than n-dodecyl-β-D-maltoside and subjected to BN-PAGE/immunoblotting. In NDUFAF4-deficient patient fibroblasts, the lack of CI led to a decrease of the I/III2- and I/III/IVn-supercomplexes (Figure 2a–c) with increased levels of CIII-dimers and the III2/IV-supercomplexes (Figure 2b and c). Of note, monomeric CIV levels remained unchanged (Figure 2c).

Complex I deficiency in patient fibroblasts carrying the NDUFAF4 variant causes specifically altered supercomplex stoichiometry. Non-denaturing BN-PAGE electrophoresis/immunoblotting analysis of mitochondrial protein (lysed with the mild detergent digitonin) from control (Ctrl) and patient (Pat) fibroblasts either transfected with wild-type NDUFAF4 (+AF4) or mock transfected with GFP (+GFP). Antibody staining was performed to detect (a) complex I (NDUFS3), (b) complex III (holo-CIII) and (c) complex IV (COX4), respectively. 'Holo-CIII' refers to an antibody raised against bovine holo-complex III that detects several different complex III subunits. Detection of complex II (SDHA) served as loading control. Complex I deficiency leads to clearly reduced abundance of the I/III2- and the I/III2/IVn- supercomplexes (SC) and monomeric complex I (CIM) that is rescued by lentiviral complementation with NDUFAF4. NDUFS3 containing subassemblies (sa) are faintly detectable in control fibroblasts. In patient fibroblasts, complex III dimers (CIII2) and supercomplexes consisting of a complex III dimer and complex IV (CIII2/CIV) are increased. Previously described uncharacterized complex IV subunit containing entities13 (#) behave alike. The levels of monomeric complex IV (CIVM) remain unchanged.
NDUFAF4 deficiency leads to accumulation of CI assembly intermediates containing constituents of the proximal P-module, the distal P-module and the N-module
We analyzed the effects of NDUFAF4 deficiency on CI assembly by 2D-BN-PAGE/SDS-PAGE experiments in patient fibroblasts (Figure 3). In the first dimension BN-PAGE, native protein complexes (that is, OXPHOS complexes) are separated (Figure 1b). The second dimension SDS-PAGE step allows separation and improved epitope availability for immunodetection of the individual components of these native protein complexes, including CI assembly intermediates. In these experiments, we used antibodies directed against representative constituents of assembly intermediates of all functional modules and, therefore, of all assembly routes of CI (Supplementary Figure 2). The Q-module subassemblies containing NDUFAF4 or NDUFS3 were not detectable anymore in patient fibroblasts and did not accumulate. However, there was a clear accumulation of proximal P-module subassemblies containing assembly factors ACAD9 and ECSIT, of distal P-module subassemblies containing CI subunit NDUFB11 and of an N-module subassembly containing CI subunit NDUFV2. These assembly defects were rescued in patient fibroblasts complemented with wild-type NDUFAF4.

Patient fibroblasts carrying the NDUFAF4 variant exhibit accumulation of complex I assembly intermediates containing parts of the proximal P-module, the distal P-module and the N-module but not the Q-module. 2D-BN-PAGE/SDS-PAGE and subsequent immunoblotting analysis of mitochondrial protein (lysed with n-dodecyl-β-d-maltoside) from control (Ctrl) and patient (Pat) fibroblasts either transfected with wild-type NDUFAF4 (+AF4) or mock transfected with GFP (+GFP). (a) The upper panel demonstrates simultaneous staining with antibodies against NDUFS3, which is a component of all Q-module subassemblies and fully assembled complex I (CI), and proximal P-module assembly factors ACAD9 and ECSIT. The lower panel demonstrates staining with an antibody against NDUFAF4. In control fibroblasts several NDUFS3 containing Q-module subassemblies (saQ) are detectable. NDUFAF4 is most strongly detectable in a low molecular weight Q-module subassembly (saQ2) but also faintly as a 'smear' reaching into the high molecular weight area (Ctrl+GFP, NDUFAF4). In mock-transfected patient fibroblasts, Q-module subassemblies are not detectable and do not accumulate: subassembly saQ1 is clearly detectable in control fibroblasts and faintly visible in NDUFAF4-transfected patient fibroblasts but not in mock-transfected patient fibroblasts. There is an accumulation of a subassembly containing proximal P-module subassembly components ACAD9 and ECSIT (saPP). Note that the signal intensity of NDUFS3 at the height of fully assembled complex I (CI) is severely reduced in mock-tranfected patient fibroblasts (Pat+GFP) while the signal intensities of both ACAD9 and ECSIT increase. The arrow indicates the faint signal of the ECSIT antibody in the panel of the mock-transfected control fibroblasts. This ratio is reversed in patient fibroblasts complemented with NDUFAF4 (Pat+AF4). (b) Staining with an antibody against distal P-module subunit, NDUFB11 reveals accumulation of two subassemblies (saPD2 and saPD3). Note that, in mock-transfected patient fibroblasts (Pat+GFP), their abundance is higher than that of high molecular weight subassembly saPD1. This ratio is reversed is patient fibroblasts transfected with wild-type NDUFAF4 (Pat+AF4). (c) Staining with an antibody against N-module subunit, NDUFV2 reveals accumulation of a subassembly (saN2). Note that, in mock-transfected patient fibroblasts (Pat+GFP), its abundance is higher than that of subassembly saN1. This ratio is reversed is patient fibroblasts transfected with wild-type NDUFAF4 (Pat+AF4).
NDUFAF4 is closely associated with NDUFAF3,1, 6 another CI assembly factor whose deficiency leads to a similar assembly defect.7 Since NDUFAF3 has been hypothesized to be involved in membrane insertion of P-module subunits,6 we analyzed the distribution of several CI subunits in mitochondrial soluble protein fractions and membrane protein fractions of patient fibroblasts (Supplementary Figure 3). In control and patient fibroblasts, P-module subunits were detectable in the membrane fraction but not in the soluble fraction.
Discussion
Prior to our report, only one NDUFAF4 variant (c.194T>C; p.(Leu65Pro)) had been reported in one family with several patients who developed encephalopathy and lactic acidosis shortly after birth.2 The patients either died in the first week of life due to intractable acidosis or developed severe myopathy, impaired neurological development and recurrent metabolic acidosis. One patient’s brain MRI at the age of 16 months revealed severe generalized brain atrophy and demyelination. The patients who survived the neonatal period passed away between the age of 9 and 18 months due to intractable acidosis.
The patient described in the present manuscript carried a novel missense NDUFAF4 variant and presented with Leigh syndrome and developmental delay. This underlines the neurological phenotype caused by NDUFAF4 deficiency while adding Leigh syndrome, a defined mitochondrial disease,8 to its spectrum.
Even though complex I deficiency can be accompanied by deficiencies of other OXPHOS complexes,9 CIII and CIV are generally stable in the case of complex I deficiency.10 However, their distribution in this setting has not been studied in patient fibroblasts. We demonstrated that non-complex I-associated CIII and CIV species do not all increase in an equal manner but are specifically altered when the formation of the I/III- and I/III/IVn-supercomplexes is hampered.
The consequences of NDUFAF4 variants for CI assembly in patient fibroblasts have previously not been studied.2, 6, 11 Our recently published detailed human CI assembly model has shown that the CI assembly process mirrors its modular architecture:1 subunits of the different functional modules are assembled separately and most intermodular association does not take place until the final steps of CI biogenesis. NDUFAF4 is associated with assembly intermediates of the Q-module.1 This association is established during early CI assembly and persists until the last steps of biogenesis, where NDUFAF4 dissociates just before completion of CI assembly.
Fractionation experiments revealed no P-module subunits in the soluble fraction and, thus, no evidence of disturbed membrane insertion in patient fibroblasts. However, previous studies have shown that P-module subunit ND1 is degraded more rapidly in the case of NDUFAF4 deficiency.12 Thus, this mechanism cannot be completely excluded, since disturbed membrane integration followed by immediate subunit degradation is a possibility.
We demonstrated accumulation of parts of all modules except for the Q-module, that NDUFAF4 associates with. The N- and P-module accumulation could be a secondary effect caused by hampered Q-module assembly. However, it could also suggest that NDUFAF4 is required to connect the separately assembled components of the P-module and the N-module to the Q-module. NDUFAF4 is most likely required for two assembly steps: (i) the connections of the PP-b/PD-a- and Q/PP-a-subassemblies to form the Q/P-subassembly1 and (ii) the addition of the N-module to complete CI (Supplementary Figure 2).
In conclusion, our study expands the phenotypic spectrum associated with NDUFAF4 deficiency and sheds light into the role of NDUFAF4 in CI assembly. Future studies will focus on the molecular mechanism of action of assembly factors.
References
Guerrero-Castillo S, Baertling F, Kownatzki D et al: The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab 2017; 25: 128–139.
Saada A, Edvardson S, Rapoport M et al: C6ORF66 is an assembly factor of mitochondrial complex I. Am J Hum Genet 2008; 82: 32–38.
Schwarz JM, Cooper DN, Schuelke M, Seelow D : MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014; 11: 361–362.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Kumar P, Henikoff S, Ng PC : Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Saada A, Vogel RO, Hoefs SJ et al: Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. Am J Hum Genet 2009; 84: 718–727.
Baertling F, Sanchez-Caballero L, Timal S et al: Mutations in mitochondrial complex I assembly factor NDUFAF3 cause Leigh syndrome. Mol Genet Metab 2016; 120: 243–246.
Baertling F, Rodenburg RJ, Schaper J et al: A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry 2014; 85: 257–265.
Saada A, Edvardson S, Shaag A et al: Combined OXPHOS complex I and IV defect, due to mutated complex I assembly factor C20ORF7. J Inherit Metab Dis 2012; 35: 125–131.
Schagger H, de Coo R, Bauer MF, Hofmann S, Godinot C, Brandt U : Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J Biol Chem 2004; 279: 36349–36353.
Marcus D, Lichtenstein M, Saada A, Lorberboum-Galski H : Replacement of the C6ORF66 assembly factor (NDUFAF4) restores complex I activity in patient cells. Mol Med 2013; 19: 124–134.
Zurita Rendon O, Shoubridge EA : Early complex I assembly defects result in rapid turnover of the ND1 subunit. Hum Mol Genet 2012; 21: 3815–3824.
Hornig-Do HT, Tatsuta T, Buckermann A et al: Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. EMBO J 2012; 31: 1293–1307.
Acknowledgements
We thank the mitochondrial diagnostics group (muscle lab, cell culture lab and DNA lab) of the Radboud Center for Mitochondrial Medicine (RCMM) at the Translational Metabolic Laboratory, Radboud UMC for excellent technical assistance. We would like to acknowledge the Genome Technology Center at the Radboud UMC and BGI Copenhagen for providing the exome sequencing service. This project was funded by the European Commission (FP7-PEOPLE-ITN. GA. 317433). Part of this work was financed by a grant obtained from the United Mitochondrial Disease Foundation (UMDF). FB was supported by a fellowship of the German Research Foundation/Deutsche Forschungsgemeinschaft (BA 5758/1-1).
Informed consent
Informed consent for diagnostic and research studies was obtained for all subjects in accordance with the Declaration of Helsinki and following the regulations of the local medical ethics committee.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Baertling, F., Sánchez-Caballero, L., van den Brand, M. et al. NDUFAF4 variants are associated with Leigh syndrome and cause a specific mitochondrial complex I assembly defect. Eur J Hum Genet 25, 1273–1277 (2017). https://doi.org/10.1038/ejhg.2017.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.133
This article is cited by
-
Multiomics analysis identifies oxidative phosphorylation as a cancer vulnerability arising from myristoylation inhibition
Journal of Translational Medicine (2024)
-
Protein lipidation in cancer: mechanisms, dysregulation and emerging drug targets
Nature Reviews Cancer (2024)
-
The host cellular protein Ndufaf4 interacts with the vesicular stomatitis virus M protein and affects viral propagation
Virus Genes (2021)
-
Molecular basis of Leigh syndrome: a current look
Orphanet Journal of Rare Diseases (2020)

{kind=link}
{kind=link}
{kind=link}