
Abstract
SeSAME/EAST syndrome is a multisystemic disorder in humans, characterised by seizures, sensorineural deafness, ataxia, developmental delay and electrolyte imbalance. It is exclusively caused by homozygous or compound heterozygous variations in the KCNJ10 gene. Here we describe a similar syndrome in two families belonging to the Malinois dog breed, based on clinical, neurological, electrodiagnostic and histopathological examination. Genetic analysis detected a novel pathogenic KCNJ10 c.986T>C (p.(Leu329Pro)) variant that is inherited in an autosomal recessive way. This variant has an allele frequency of 2.9% in the Belgian Malinois population, but is not found in closely related dog breeds or in dog breeds where similar symptoms have been already described. The canine phenotype is remarkably similar to humans, including ataxia and seizures. In addition, in half of the dogs clinical and electrophysiological signs of neuromyotonia were observed. Because there is currently no cure and treatment is nonspecific and unsatisfactory, this canine translational model could be used for further elucidating the genotype/phenotype correlation of this monogenic multisystem disorder and as an excellent intermediate step for drug safety testing and efficacy evaluations before initiating human studies.
Similar content being viewed by others

Introduction
SeSAME (Seizures, Sensorineural deafness, Ataxia, Mental retardation and Electrolyte imbalance) or EAST (Epilepsy, Ataxia, Sensorineural deafness, Tubulopathy) syndrome (Phenotype MIM number 612780) is a rare, autosomal recessive multisystemic disorder.1, 2 It is caused by homozygous or compound heterozygous variations in the voltage-gated potassium channel (VGKC), inwardly rectifying, subfamily J, member 10 gene (KCNJ10, alias KIR4.1; Gene/Locus MIM number 602208). Functional KCNJ10 subunits form homotetrameric or heterotetrameric (in coassembly with KCNJ16, alias KIR5.1) channels with two putative transmembrane regions. The KCNJ10 channel is mainly expressed in the brain, spinal cord, inner ear and kidneys. Depending on their tissue localisation, these channels have distinct physiologic properties. Variations described so far have all been loss-of-function variations.3
The human KCNJ10 gene is very well characterised. It is annotated in all genome browsers, has a reviewed RefSeq status (NCBI Gene ID 3766) and one transcript (CCDS1193.1; annotated by both Ensembl and HAVANA). It is located on chromosome 1q23.2 and consists of a first noncoding exon and a second exon containing the complete CDS of 1140 bp encoding a protein of 379 amino acids. Until now, 14 variants have been described causing SeSAME/EAST, all in the KCNJ10 coding sequence (Figure 1).4

A similar disease (OMIA 001820-9615) has also been described in a number of dog breeds (Border collie, Dachshund and Terrier breeds; reviewed in Vanhaesebrouck et al5). Until now, only the KCNJ10 c.627C>G (p.(Ile209Met)) variant has been reported to be associated with spinocerebellar ataxia and myokymia, seizures or both (SAMS) in certain Terrier breeds.6, 7 In addition, Forman et al8 reported that the CAPN1 c.344G>A (p.(Cys115Tyr)) variant is strongly associated with late-onset ataxia in Parson Russell Terriers, suggesting that CAPN1 may represent a novel candidate gene for ataxia in humans. According to OMIA there are no spontaneous disease causing variants in KCNJ10 described in other species.
Here, we describe a syndrome in two Malinois dog families with a phenotype strikingly similar to SeSAME/EAST syndrome in humans and the identification of a new pathogenic missense variant in KCNJ10.
Materials and methods
Clinical examination
Three 3-month-old intact (two males and one female) Malinois dogs (dogs 1–3) were presented at the Department of Small Animal Medicine and Clinical Biology, Faculty of Veterinary Medicine, Ghent University, for an uncoordinated gait since the age of 6 to 8 weeks. A clinical and neurological examination was performed in all three dogs, and a complete bloodwork in dogs 1 and 3. Urinalysis was performed only in dog 3. Electromyography (EMG) and brainstem auditory evoked response (BAER) tests were performed in all three dogs. EMG recordings were made from facial, truncal and appendicular muscles of front and hind limbs. Motor nerve conduction velocity studies and repetitive nerve stimulation of the radial and sciatic nerves were performed in dogs 1 and 2. A commercially available electrophysiological unit (Medelec/TECA, Sapphire 2M, Surrey, UK) was used for electrodiagnostic recordings. EMG and BAER tests were performed under sedation and motor nerve conduction velocity studies were performed under general anaesthesia. Cerebrospinal fluid analysis and MRI (0.2 Tesla magnet) of the brain and cervical spinal cord was performed in dog 1. For affected dog 4, only video footage was available.
Pathological examination
Post-mortem examination was performed in dog 2, immediately following the killing. Central and peripheral nervous tissue samples were collected, formalin-fixed, paraffin-embedded and stained with haematoxylin–eosin or luxol fast blue. An immunohistochemical staining with neurofilament (monoclonal mouse anti-human neurofilament protein clone 2F11; Cat. No. M0762, Dako, Glostrup, Denmark) was performed.
Genetic analysis
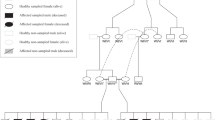
EDTA blood samples from the four affected dogs and nine healthy family members were collected for genetic analysis (pedigree analysis in Figure 2). Genomic DNA was isolated from the blood by performing a proteinase K digestion as described in Van Poucke et al.9 cDNA transcribed from canine brain RNA in Van Poucke et al10 was used in this study to experimentally investigate the existence of the three predicted KCNJ10 transcript variants (Acc. No.: X1: XM_005640901.2, X2: XM_014111261.1, X3: XM_545752.5).

Sample pedigree of the 2 families (family 1 at the left and family 2 at the right) drawn with Madeline 2.0 PDE.50 Squares are males and circles females. The white shaded icon represents a not blood-sampled dog, grey were healthy dogs and black were affected dogs. Strikethrough icons represent deceased animals. The KCNJ10 c.986T>C genotype is shown underneath the icon.
Primer pairs were designed with Primer-BLAST11 based on the canine KCNJ10 reference sequence (Acc. No. NC_006620.3), taking into account all described transcript variants, sequence variants and repeat sequences. Primers were chosen in regions that were free of secondary structures (Mfold).12 PCR amplicons were analysed via agarose gel electrophoresis. Sequencing reactions were performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) with the individual PCR primers as sequencing primers and run at Eurofins Genomics (Ebersberg, Germany). Sequence analysis was performed with Unipro UGENE v1.16.1.13 An assay with dual-labelled probes (Eurofins) was designed as described by Van Poucke et al9 to genotype the KCNJ10 c.986T>C variant in additional animals. Experimental details on the genetic analysis are given in Supplementary Information File S1A.
Bioinformatics tools for sequence variant interpretation
Variant information was gathered from population databases (Exome Aggregation Consortium14, Exome Variant server15, 1000 Genomes16, dbSNP17 and dbVAR18), disease databases (humsavar.txt release 2016_0519, ClinVar20, HGMD-public21, OMIM22 and OMIA23) and sequence databases (RefSeqGene24, NCBI Genome25, UCSC Genome26 and Ensembl Genome27). I-Mutant2.028 was used as a predictor of protein stability changes upon variations. Computational prediction of disease-related variants was performed with the consensus tools CONDEL29 (combines Logre, MAPP, Massessor, Pph2 and SIFT), Meta-SNP30 (combines PANTHER, PhD-SNP, SIFT and SNAP), PredictSNP31 (combines MAPP, PhD-SNP, Pph1, Pph2, SIFT and SNAP) and PON-P2.32
Results
Clinical features
General clinical examination was unremarkable in all three presented dogs. Neurological examination revealed a severe generalised hypermetric ataxia and absent patellar reflexes in all dogs (Supplementary Information File S2A). Generalised involuntary vermicular muscle contractions (myokymia) triggered by excitement were present in dogs 1 and 2 (Supplementary Information File S2B). Dog 1 had demonstrated a neuromyotonic episode of extreme generalised muscle stiffness with normal consciousness, triggered by stress. The dog also developed generalised seizures a few weeks later.
Complete blood count and serum biochemistry, including electrolytes (Na+, K+, Cl-, Ca2+ and Mg2+), was normal in dogs 1 and 3. Urinalysis (including electrolyte clearance for Na+, K+, Cl-, Ca2+, Mg2+ and PO4-), was unremarkable in dog 3. EMG showed neuromyotonic discharges5, 33 in those muscles also visibly showing myokymia in dogs 1 and 2 (Figure 3), and was normal in dog 3. BAER recordings showed disappearance of wave components III/IV and V, and mildly prolonged latencies of waves I and II in dogs 1 to 3 (Figure 4). Results of motor nerve conduction velocity studies and repetitive nerve stimulation in dogs 1 and 2 did not differ from two age-matched Malinois control dogs. MRI of the brain/cervical spine and cerebrospinal fluid analysis, performed in dog 1, did not reveal any abnormalities.

EMG: neuromyotonic discharges in a muscle clinically affected with myokymia. Multiple waveforms consisting of trains of 2 to 6 motor unit action potentials.

BAER potentials of an affected (A) and a control (C) dog (1 ms/division): loss of waves III/IV and V, prolonged latencies of all waves and prolonged interpeak latencies.
The neurological condition of dogs 1, 2 and 4 deteriorated further (Supplementary Information File S2C) and all were killed because of the severity of the ataxia before the age of 6 months. Despite severely debilitating non-ambulatory ataxia, dog 3 is still alive (9 months after presentation).
Pathological features
A full necropsy was performed on dog 2 (ataxia and myokymia). No macroscopic abnormalities were found. On histopathology, bilateral myelopathy with predominant axonopathy and myelin vacuolisation was found. Changes were most obvious in the corticospinal tracts of the brainstem and the ventral funiculi of the cervical, thoracic and lumbar spinal cord. Similar lesions were found in the cerebellum (white matter) and medulla oblongata (predominantly at the pyramid bodies). In the peripheral nervous system, subtle changes were seen in a few axons (swollen with vacuolated myelin) of the brachial plexus, the femoral nerve and the sciatic nerve. Luxol fast blue and neurofilament stainings confirmed these findings (Supplementary Information File 3).
Screening for known spinocerebellar ataxia-related/causal variants
All 13 members of the 2 Malinois families (dogs 1–13, Figure 2) were first screened for the KCNJ10 c.627C>G (p.(Ile209Met)) variant (Acc. No. XM_545752.3) and the CAPN1 c.344G>A (p.(Cys115Tyr)) variant (Acc. No. XM_540866.3), as performed by Gilliam et al,6 but none of the variants (so far only described in certain Terrier breeds) were detected. In addition, none of the variants were found in 28 healthy Malinois from different families.
Canine KCNJ10 gene characterisation
Contrary to the human KCNJ10, its canine orthologue is not yet annotated in any of the genome browsers (although assigned to chromosome 38), only has a model RefSeq status (NCBI Gene ID 488635) and three predicted transcript variants (X1-X3, with X3 being orthologous with the human transcript variant). Therefore, we first validated the predicted canine KCNJ10 transcripts experimentally. Via RT-PCR we could only confirm the presence of transcript variant X3 in the canine brain (primers 1F/1R could only amplify the 360-bp X3 fragment and not the 535-bp X1 fragment whereas primers 2F/R2 could not amplify the 140-bp X2 fragment, although they could amplify a 231-bp fragment with genomic DNA as a template; see Supplementary Information File S1A for experimental details). The other two canine transcript variants were not or very lowly (below detection limit) expressed in the canine brain, or wrongly predicted. Sequencing the complete X3 transcript showed that the canine KCNJ10 CDS was correctly predicted and has the same length compared with its human transcript orthologue, showing 90% nucleotide sequence identity and 98% amino acid sequence identity (Supplementary Information File S1B–C).
KCNJ10 variant detection
The complete confirmed canine KCNJ10 CDS was resequenced in both parent dogs of family 1, their 5 offspring (including 2 affected dogs) and one healthy Malinois from another family. Only one variant was discovered, a c.986T>C (p.(Leu329Pro)) variant (NCBI_ss#1966650805), homozygous in the 2 affected offspring (dogs 1–2), heterozygous in both healthy parents (dogs 10–11) and 2 healthy offspring (dogs 6–7), and not present in 1 healthy offspring (dog 8) and 1 non-related healthy Malinois (Figure 2 and Supplementary Information File S1B–D).
KCNJ10 variant screening
A genotyping assay with dual-labelled probes was used to screen this variant in another healthy offspring of the father in family 1 (dog 5, heterozygous) and in family 2 (healthy heterozygous parents (dogs 12–13), 2 affected homozygous mutant offspring (dogs 3 and 4) and 1 healthy heterozygous offspring (dog 9; Figure 2). The assay was also used to estimate the frequency of this variant in 103 additional healthy Malinois from different families: 97 were homozygous wild type, 6 heterozygous and none were homozygous mutant. We also investigated whether the variant was present in the other closely related Belgian Shepherd breeds (Groenendaeler (N=52), Laekenois (N=28) and Tervueren (N=56)), other Sheepdogs (Australian shepherd, German shepherd and Shetland sheepdog; all N=25) and breeds where similar symptoms have been already described (Border collie, Dachshund, Jack Russell terrier, Maltese and Yorkshire terrier; all N=25). However, the variant was not detected in any of these breeds.
Discussion
Based on the Standards and Guidelines for the Interpretation of Sequence Variants, we conclude that the c.986T>C (p.(Leu329Pro)) variant is pathogenic for the SeSAME/EAST-like syndrome in Malinois dogs in an autosomal recessive way because of 1 strong (PS), 1–2 moderate (PM) and 3–4 (PP) supporting criteria for pathogenicity.34
All 4 affected Malinois dogs were homozygous mutant, whereas none of the 112 healthy Malinois dogs were. Because there were no false negatives/positives, odds ratios were infinite (PS4). Apart from the 13 members of the affected families (Figure 2), only 6 carriers were observed in the other 103 tested healthy Malinois dogs. The variant was not found in 136 dogs from 3 closely related Belgian breeds, 75 dogs from 3 other sheepdog breeds and 125 dogs from 5 breeds where similar symptoms have been already described (all dogs came from different families). In addition, a Leu was present at this position in the intracellular C-terminus in all known eutherian mammal KCNJ10 reference sequences and a variant in this codon was not found in popular population databases. A variation in this highly conserved residue is assumed to be pathogenic (PM2).
In humans, SeSAME/EAST is exclusively caused by variants in KCNJ10. Until now, 14 different pathogenic variants have been described with functional confirmation (Figure 1). A KCNJ10 missense variant was also found to segregate with a similar disease in certain Terrier breeds. Here we describe a novel KCNJ10 missense variant that segregates with the disease in two Malinois families (with two affected family members in each family; PM/PP1). As the same variation segregates in both families, they probably share a common ancestor carrying the founder variation. However, because of the lack of pedigree information, we could not identify the founder or the relatedness between both families. The allele frequency of the variant in the Belgian Malinois population is estimated at 2.9%. Breeders are advised to screen for the variant in order to breed with variant-free animals and get variant-free offspring.
Missense variants are the most common mechanism in this disease (11/14 in humans and 2/2 in dogs) and missense variants in KCNJ10 have a low rate of benignity (z-score for missense variants in KCNJ10 is 1.78 according to the ExAC Browser, indicating intolerance to variation; PP2).
There are multiple lines of computational evidence that support the deleterious effect of this Leu→Pro variant. First, the introduction of a proline is known to affect the protein stability because of its exceptional structure. It is conformationally more rigid than any other amino acid because of its pyrrolidine ring and unlike all other amino acids it contains a secondary amide (instead of a primary amide) being unable to form a hydrogen bond.35 According to I-Mutant2.0, the p.(Leu329Pro) variation indeed decreases stability (RI=8). Second, according to humsavar.txt (release 2016_05) 65% of all Leu→Pro variants are disease associated, whereas the average for all variants is only 38%. Leu→Pro variants are together with Gly→Arg and Arg→Cys variants the most predominant disease-associated variants (all more than 1000 hits). In addition, all nine classified Leu→Pro variants known to exist in the KCN gene family are disease associated. Third, frequently used consensus tools for computational prediction of disease-related variants all confirm this assumption (CONDEL: 0.55, deleterious; Meta-SNP: 0.68, disease; PredictSNP: 0.55, deleterious; PON-P2: 0.91, pathogenic). Finally, the patients’ phenotype and family history is highly specific for a disease with a single genetic aetiology (PP4).
The clinical features of the SeSAME/EAST syndrome are the consequence of a KCNJ10 potassium channel dysfunction.1, 36 In our four affected dogs, ataxia was always present, with or without myokymia, neuromyotonia and/or seizures. It is believed that, as in humans, the novel KCNJ10 variant affects the function of the potassium channel in the brain and spinal cord, resulting in seizures and ataxia in the affected Malinois dogs.
Interestingly, clinical signs of myokymia and neuromyotonia (nor cramps) have not been described in humans and also not in knockout KCNJ10 mice models. Myokymia and neuromyotonia were clinically and electrodiagnostically detected in respectively 2 and 1 of the affected Malinois dogs. The authors believe that the association between myokymia/neuromyotonia and KCNJ10 variation is not coincidental, as another KCNJ10 variation also caused myokymia and neuromyotonia in a large number of dogs.6, 7 In humans, myokymia and neuromyotonia have been associated with genetic and immune-mediated defects of voltage-gated potassium channels of the peripheral nerve.37, 38, 39, 40, 41, 42 However, it has not yet been associated with a potassium channel in the central nervous system. One hypothesis would be that impairment of the ‘potassium sink’ role by a KCNJ10 variation not only can result in increased neuronal firing within the brain and resulting in seizures, but also in the lower motor neurons of the spinal cord, resulting in hyperexcitability of the peripheral nerve and consequently resulting myokymia and/or neuromyotonia. Another hypothesis would be that the KCNJ10 channel is also present in the peripheral nerve itself, as suggested in literature, but this has not been investigated yet. Moreover, next to direct malfunctioning of potassium channels, demyelination of the peripheral nerve has also been reported together with myokymia and neuromyotonia, another hypothesis in our animal model as demyelination of peripheral nerves was observed. The authors do not believe that systemic electrolyte disturbances would have resulted in myokymia and neuromyotonia in the affected Malinois dogs, as serum electrolytes and electrolyte clearance were normal. The fact that all dogs had ataxia, but only some dogs had seizures, myokymia or neuromyotonia, is not surprising, as it is well known that potassium channel diseases can be phenotypically heterogeneous (variability of clinical signs and severity) in dogs33 and humans.1, 2, 43
The mildly delayed latencies of waves I and II, and the loss of waves III, IV and V, was repetitively found on the BAER tests of our affected dogs. This suggests a peripheral as well as central localisation for dysfunction within the auditory pathway.44 This is a major difference in humans and mice, in which a cochlear dysfunction is assumed to be the origin of the hearing impairment.1, 2 The extent of hearing impairment in humans with KCNJ10 variation is variable and can sometimes be absent or only appreciated with specific testing.45 None of the affected dogs showed any signs of clinically relevant hearing impairment.
Another major difference with the SeSAME/EAST syndrome in humans is that the dogs in this study did not appear to show any signs of developmental delay as, apart from gait abnormalities, they behaved and were able to obey as well as their healthy siblings. Because intelligence and language testing as in humans are not applicable to dogs, we cannot exclude that a certain degree of developmental delay could have been missed in our dogs.
The histopathological findings in our dogs are identical to the ones described in the Russel Terrier group.6, 8 In knockdown mice, hypomyelination in the spinal cord with severe spongiform vacuolation, axonal swelling and degeneration is also described.46 To the author’s knowledge, there are currently no pathological reports available of humans with SeSAME/EAST syndrome. A nerve biopsy report from a human patient showed axonal neuropathy with hypomyelination, consistent with the findings in dogs.45
In humans, the ataxia seems to be nonprogressive and the seizures are in most cases easily controlled with antiepileptic drugs.43 In mice, general knockout of KIR4.1 leads to neonatal mortality.47 In Jack Russel Terriers, the ataxia also seems to be nonprogressive, but the myokymia and neuromyotonia worsen gradually over several months, leading to killing of most dogs.33 Because of the larger stature of Malinois dogs compared with Jack Russell Terriers, they seemed to cope less easily with the ataxia during growth of the animal. Hence, mid- to long-term prognosis in these dogs is reserved to poor.
In conclusion, we described a pathogenic c.986T>C (p.(Leu329Pro)) variant for a SeSAME/EAST-like syndrome in Malinois dogs. Because, currently, there is no cure and treatment is nonspecific and unsatisfactory, the canine translational model could, in addition to the existing valuable model organisms such as Xenopus, mice and zebrafish that are mainly used for basic research and drug screening,1, 48 be used for further elucidating the genotype/phenotype correlation of this monogenic multisystem disorder and as an excellent intermediate step for drug safety testing and efficacy evaluations before initiating human studies.49
References
Bockenhauer D, Feather S, Stanescu HC et al: Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. New Engl J Med 2009; 360: 1960–1970.
Scholl UI, Choi M, Liu T et al: Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 2009; 106: 5842–5847.
Sala-Rabanal M, Kucheryavykh LY, Skatchkov SN, Eaton MJ, Nichols CG : Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10). J Biol Chem 2010; 285: 36040–36048.
Parrock S, Hussain S, Issler N et al: KCNJ10 mutations display differential sensitivity to heteromerisation with KCNJ16. Nephron Physiol 2013; 123: 7–14.
Vanhaesebrouck AE, Bhatti SF, Franklin RJ, Van Ham L : Myokymia and neuromyotonia in veterinary medicine: a comparison with peripheral nerve hyperexcitability syndrome in humans. Vet J 2013; 197: 153–162.
Gilliam D, O’Brien DP, Coates JR et al: A homozygous KCNJ10 mutation in Jack Russell terriers and related breeds with spinocerebellar ataxia with myokymia, seizures, or both. J Vet Intern Med 2014; 28: 871–877.
Rohdin C, Gilliam D, O'Leary CA et al: A KCNJ10 mutation previously identified in the Russell group of terriers also occurs in Smooth-Haired Fox Terriers with hereditary ataxia and in related breeds. Acta Vet Scand 2015; 57: 26.
Forman OP, De Risio L, Mellersh CS : Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the Parson Russell Terrier dog breed. PLoS One 2013; 8: e64627.
Van Poucke M, Vandesompele J, Mattheeuws M, Van Zeveren A, Peelman LJ : A dual fluorescent multiprobe assay for prion protein genotyping in sheep. BMC Infect Dis 2005; 5: 13.
Van Poucke M, Martlé V, Van Brantegem L et al: A canine orthologue of the human GFAP c.716G>A (p.Arg239His) variant causes Alexander disease in a Labrador retriever. Eur J Hum Genet 2016; 24: 852–856.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden T : Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 2012; 13: 134.
Zuker M. : Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003; 31: 3406–3415.
Okonechnikov K, Golosova O, Fursov M, UGENE team: Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 2012; 28: 1166–1167.
Exome Aggregation Consortium (ExAC). Cambridge, MA. Available from http://exac.broadinstitute.org/ (accessed May 2016).
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available from http://evs.gs.washington.edu/EVS/ (accessed May 2016).
The 1000 Genomes Project Consortium: A global reference for human genetic variation. Nature 2015; 526: 68–74.
Sherry ST, Ward MH, Kholodov M et al: dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001; 29: 308–311.
Lappalainen I, Lopez J, Skipper L : dbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res 2013; 41: D936–D941.
Wu CH, Apweiler R, Bairoch A et al: The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res 2006; 34: D187–D191.
Landrum MJ, Lee JM, Benson M et al: ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016; 44: D862–D863.
Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN : The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014; 133: 1–9.
Online Mendelian Inheritance in Man, OMIM. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University: Baltimore, MD. Available from http://omim.org/ (accessed May 2016).
Online Mendelian Inheritance in Animals, OMIA. Faculty of Veterinary Science, University of Sydney: Sydney. Available from http://omia.angis.org.au/ (accessed May 2016).
Pruitt KD, Brown GR, Hiatt SM et al: RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 2014; 42: D756–D763.
NCBI Genome. Available from http://www.ncbi.nlm.nih.gov/genome/ (accessed May 2016).
Kent WJ, Sugnet CW, Furey TS et al: The human genome browser at UCSC. Genome Res 2002; 12: 996–1006.
Yates A, Akanni W, Amode MR et al: Ensembl 2016. Nucleic Acids Res 2016; 44: D710–D716.
Capriotti E, Fariselli P, Casadio R : I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 2005; 33: W306–W310.
González-Pérez A, López-Bigas N : Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 2011; 88: 440–449.
Capriotti E, Altman RB, Bromberg Y : Collective judgment predicts disease-associated single nucleotide variants. BMC Genomics 2013; 14: S2.
Bendl J, Stourac J, Salanda O et al: PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 2014; 10: e1003440.
Niroula A, Urolagin S, Vihinen M : PON-P2: prediction method for fast and reliable identification of harmful variants. PLoS One 2015; 10: e0117380.
Bhatti SF, Vanhaesebroeck AE, Van Soens I et al: Myokymia and neuromyotoni in 37 Jack Russel terriers. Vet J 2011; 189: 284–288.
Richards S, Aziz N, Bale S et al: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424.
Bajaj K, Madhusudhan MS, Adkar BV et al: Stereochemical criteria for prediction of the effects of proline mutations on protein stability. PLoS Comput Biol 2007; 3: e241.
Reichold M, Zdebik AA, Lieberer E et al: KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci USA 2010; 107: 14490–14495.
Hart IK, Newsome-Davis J (eds): Generalized Peripheral Nerve Hyperexcitability (Neuromyotonia). New York, USA: Mc-Graw-Hill, 2004.
Browne DL, Gancher ST, Nutt JG et al: Episodic ataxia/myokymia syndrome associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 1994; 8: 136–140.
Wuttke TV, Jurkat-Rott K, Paulus W, Garncarek M, Lehmann-Horn F, Lerche H : Peripheral nerve hyperexcitability due to dominant-negative KCNQ2 mutations. Neurology 2007; 69: 2045–2053.
Irani SR, Alexander S, Waters P et al: Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133: 2734–2748.
Kleipa KA, Elman LB, Lang B, Vincent A, Scherer SS : Neuromyotonia and limbic encephalitis sera target mature Shaker-type K+ channels: subunit specificity correlates with clinical manifestations. Brain 2006; 129: 1570–1584.
Odabasi Z, Joy JL, Claussen GC, Herrera GA, Oh SJ : Isaac’s syndrome associated with chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 1996; 19: 210–215.
Cross JH, Arora C, Heckemann RA et al: Neurological features of epilepsy, ataxia, sensorineural deafness, tubulopathy syndrome. Dev Med Child Neurol 2013; 55: 846–856.
Dewey CW, da Costa RC (eds): Practical Guide to Canine and Feline Neurology, 3rd edn . Iowa, USA: Wiley Blackwell, 2016.
Scholl UI, Dave HB, Lu M et al: SeSAME/EAST syndrome – phenotypic variability and delayed activity of the distal convoluted tubule. Pediatr Nephrol 2012; 27: 2081–2090.
Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P : Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci 2001; 21: 5429–5438.
Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD : Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 2007; 27: 11354–11365.
Mahmood F, Mozere M, Zdebik AA et al: Generation and validation of a zebrafish model of EAST (epilepsy, ataxia, sensorineural deafness and tubulopathy) syndrome. Dis Model Mech 2013; 6: 652–660.
Patterson EE. : Canine epilepsy: an underutilized model. ILAR J 2014; 55: 182–186.
Trager EH, Khanna R, Marrs A et al: Madeline 2.0 PDE: a new program for local and web-based pedigree drawing. Bioinformatics 2007; 23: 1854–1856.
Acknowledgements
We thank Dominique Vander Donckt, Linda Impe and Ruben Van Gansbeke for excellent technical assistance, as well as the breeders, owners and veterinarians who collaborated to this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Van Poucke, M., Stee, K., Bhatti, S. et al. The novel homozygous KCNJ10 c.986T>C (p.(Leu329Pro)) variant is pathogenic for the SeSAME/EAST homologue in Malinois dogs. Eur J Hum Genet 25, 222–226 (2017). https://doi.org/10.1038/ejhg.2016.157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.157
This article is cited by
-
Identification and functional characterization of two novel mutations in KCNJ10 and PI4KB in SeSAME syndrome without electrolyte imbalance
Human Genomics (2019)
-
Truncating SLC12A6 variants cause different clinical phenotypes in humans and dogs
European Journal of Human Genetics (2019)
