
Abstract
Variants in family 1 kinesin (KIF1A), which encodes a kinesin axonal motor protein, have been described to cause variable neurological manifestations. Recessive missense variants have led to spastic paraplegia, and recessive truncations to sensory and autonomic neuropathy. De novo missense variants cause developmental delay or intellectual disability, cerebellar atrophy and variable spasticity. We describe a family with father-to-son transmission of de novo variant in the KIF1A motor domain, in a phenotype of pure spastic paraplegia. Structural modeling of the predicted p.(Ser69Leu) amino acid change suggested that it impairs the stable binding of ATP to the KIF1A protein. Our study reports the first dominantly inherited KIF1A variant and expands the spectrum of phenotypes caused by heterozygous KIF1A motor domain variants to include pure spastic paraplegia. We conclude that KIF1A should be considered a candidate gene for hereditary paraplegias regardless of inheritance pattern.
Similar content being viewed by others

Introduction
Kinesins are motor proteins that use ATP to power the movement of cargoes along microtubules.1 The neuron-specific family 1 kinesin (KIF1A) transports presynaptic vesicles toward the distally located plus end of the axonal microtubules.2 KIF1A contains an N-terminal motor domain, a forkhead associated domain and a C-terminal pleckstrin homology domain.3
Homozygous disruption of Kif1a in mice led to motor and sensory disturbances and to death soon after birth.4 The first disease-associated variants in KIF1A, reported in patients with hereditary sensory and autonomic neuropathy II (HSANII), were recessive truncating variants abolishing the C-terminus of the protein.5 Next, homozygous missense variants in the KIF1A motor domain were found causative for the autosomal recessive hereditary spastic paraplegia (HSP) subtype SPG30.6, 7, 8 HSP is characterized by the degeneration of axons of the corticospinal tract motor neurons. If additional symptoms or signs are present, the patient has ‘complicated’ rather than ‘pure’ HSP.9
The phenotypic spectrum of KIF1A-related disease grew recently further with the identification of de novo missense variants in the motor domain in patients who had intellectual disability (ID) in addition to variable other symptoms including spastic paraparesis, axonal neuropathy and cerebellar atrophy.10, 11, 12 Here we report the first dominantly inherited KIF1A variant, which caused pure HSP.
Materials and methods
Patients
The index patient of Finnish origin was diagnosed at the Helsinki University Central Hospital (HUCH). All participants or guardians gave written informed consent. The study was approved by the ethics board of HUCH.
Sequencing
For gene screening, a targeted next-generation sequencing (NGS) method was used that included 136 genes linked to HSP, neuropathy or related diseases (Supplementary Table 1). Target enrichment and amplification were done with the HaloPlex kit (Agilent Technologies, Santa Clara, CA, USA) and sequencing on MiSeq (Illumina, San Diego, CA, USA) as described.13 Alignment and variant calling was done as previously reported.14
Data filtering
NGS single nucleotide variant data were filtered as follows: (1) Exclusion of variants with frequency >0.005 in the 1000 genomes database (http://www.1000genomes.org/, accessed 4/2014). (2) Exclusion of synonymous or non-splice site changing variants. (3) Exclusion of some recurring false positive calls only present in reads from one restriction enzyme fragment; using Integrative Genomics Viewer.15 (4) Exclusion of common Finnish variants with frequency >0.001 in the Sequencing Initiative Suomi (SISu) database (http://sisu.fimm.fi/, accessed 4/2014).16
Insertion and deletion (indel) data were filtered by: (1) Exclusion of variants outside of genes; (2) Exclusion of synonymous or non-splice site changing; (3) Exclusion of variants found in the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP/, accessed 1/2014).
Predictions of variants’ pathogenicity were obtained using SIFT (version 1.03, http://sift.jcvi.org/),17 PolyPhen-2 (version 2.2.2r398, http://genetics.bwh.harvard.edu/pph2/)18 and MutationTaster (version 2, http://www.mutationtaster.org/)19 software.
Sanger sequencing
Sanger sequencing was used to confirm the identified variant and to test the family members. Oligonucleotide primer sequences used for the KIF1A (NG_029724.1, NM_001244008.1) variant were: 5′-CCTGCAACTTCATTCCTTCC-3′ and 5′-GCTCTCAGCCTCAGCTGGT-3′. Genetic variant data were submitted to ClinVar (accession number SCV000191086, http://www.ncbi.nlm.nih.gov/clinvar/).
Structural modeling of the KIF1A motor domain
The structural modeling was based on the crystal structure of KIF1A motor domain (PDB ID: 1VFV). The variant was introduced by using mutagenesis option in PyMOL, molecular visualization software.20 The energy minimization and loop flexible modeling were performed by using Modeller software.21 All structural images were generated using PyMOL.20
Results
Clinical findings
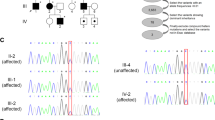
Our index patient is a 9-year-old boy (Figure 1a), who was sent to our pediatric neurology unit at the age of 2.5 years because of toe walking and spasticity in the lower extremities. His father had similar symptoms as a toddler, and cerebral palsy was suspected, but the brain imaging was normal. Since then the father has had lower-extremity spasticity with walking difficulties. The paternal grandparents have had no problems with walking. Both the patient and his father had accelerated reflexes in the lower limbs with positive Babinski sign. The diagnosis was set as both had the typical physical appearance of spastic paraparesis: a muscular trunk with lumbar lordosis, stiff lower extremities, cavus feet and spontaneous dorsal flexion of the first toe. ENMG was performed to the patient’s father with normal results. Brain and spinal MRI of the patient were normal. The patient’s cognitive development has been normal based on neuropsychological tests. The father had no learning difficulties at school, but the patient has received special education because of attention deficit disorder (ADD) since the first grade. Because of the ADD, MRI was controlled and electroencephalography (EEG) registered at the age of 8 years. The MRI showed no abnormality and EEG revealed focal frontal disturbance with normal background activity and no spikes or bursts.

(a) Pedigree of the affected family (proband highlighted with arrow) and (b) Sanger sequencing chromatograms show DNA from the proband (lower), his parents (middle) and paternal grandparents (upper). No evidence of blood DNA mosaicism was detected in the grandparents. (c) Amino acid conservation of the KIF1A motor domain region in which p.S69 is located (arrow).
Genetic findings
Filtering of the proband’s NGS data left a single heterozygous variant, c.206C>T in the KIF1A gene (NG_029724.1, NM_001244008.1), which predicted a p.(Ser69Leu) amino-acid change (Supplementary Table 2). The presence of the variant was confirmed by Sanger sequencing (Figure 1b). The variant was found to segregate with the disease phenotype, as the proband’s father carried the variant but his brother and mother were both negative as were the father’s parents. These results showed that the c.206C>T variant had arisen de novo in the father and was inherited by the proband. No signs of mosaicism were observed for the c.206C>T variant in the paternal grandparents’ blood DNA. This variant was absent in 1000 Genomes, EVS and SISu databases. The amino acid p.Ser69 is conserved in mammals and birds (Figure 1c). The variant was predicted damaging by SIFT, probably damaging by Polyphen-2 and disease causing by MutationTaster.
Structural modeling
The p.Ser69 residue is located in the motor domain of KIF1A (Figures 2a and b). The movement of KIF1A along the microtubules is powered by the hydrolysis of ATP. Structural modeling, based on the crystal structure of the KIF1A motor domain,22, 23 suggested that the p.(Ser69Leu) change, which was predicted based on the observed c.206C>T variant, might affect the conformation of the L2 loop, which participates in the formation of the ATP binding pocket. In particular, p.Tyr67 binds the adenine group of ATP in a water-mediated manner (Figure 2e) and the conformational change induced by p.Ser69Leu could break the interaction between p.Tyr67 and ATP, thus affecting the stable ATP binding of KIF1A (Figure 2f). The electrocharge distribution of the ATP binding pocket suggested that its overall structure is unlikely to be significantly affected by the predicted p.(Ser69Leu) change (Figures 2c, d, g and h).

(a, b) Overall structure of the KIF1A motor domain. Missing loops are indicated by dashed lines. The switch I, switch II, P-loop and neck linker are indicated in blue, magenta, green and yellow, respectively. The location of p.Ser69 and ATP analogue (AMP-PNP, adenylyl imidodiphosphate) are shown. (c, d) Electrocharge distribution of wild-type and p.(Ser69Leu) KIF1A. Negative and positive charges are represented by red and blue color, respectively. (e) ATP binding interface of KIF1A in crystal structure. Phosphate groups are held by several hydrogen bonds (dotted line). p.Tyr105 shows a phi–phi interaction with the adenine group from ATP. p.Tyr67 from the L2 loop shows water-mediated bonding with the N1 of adenine group. (f) Structural impact of p.(Ser69Leu) change. The substitution might affect the stability of the L2 loop and change the conformation of p.Tyr67, disrupting its binding to the adenine group of ATP. (g) Views of the KIF1A ATP binding pocket and (h) the modeled structure of p.(Ser69Leu) variant.
Discussion
The present study enlarges the spectrum of KIF1A-associated disease by describing the first dominantly inherited disease variant. Our proband was heterozygous for a previously unknown c.206C>T (p.(Ser69Leu)) variant in KIF1A. His father carried the same variant, whereas the father’s parents did not. This showed that the variant was de novo in the father and was transmitted from father to son. Heterozygous KIF1A variants had not been described in HSP phenotypes until a recent report by us and collaborators where 11 de novo variants were described in 14 patients.10 The predominant phenotype in these cases was developmental delay and/or ID with cerebellar, cerebral and/or optic nerve atrophy. Spasticity was present in most but not all patients. Disease severity varied considerably. One of the most severely affected patients was a female with a c.757G>A (p.(Glu253Lys)) variant. She had hypotonia and decreased movements at birth, examinations revealed progressive cerebral atrophy and axonal sensorimotor neuropathy, and she died at age 2. Among the least severely affected, and the only one who had reached adult age at the time of the study, was a Finnish patient with a c.305G>A (p.(Gly102Asp)) variant. She presented with ID, gradually developing spastic paraparesis but no peripheral neuropathy. The c.206C>T (p.(Ser69Leu)) variant identified in this study represents the least severe heterozygous KIF1A variant as our patients had pure HSP and normal cognitive development. Although our proband had ADD, it cannot be accounted for by the KIF1A variant as the father has not had such challenges in his school or working life. The proband also had focal frontal disturbance in EEG. We cannot definitely ascribe the EEG finding to the KIF1A variant as such unspecific, clinically insignificant abnormalities are commonly found in children who are evaluated for mild neurocognitive abnormalities. Nevertheless, epilepsy or EEG abnormalities were identified in five previously described patients with de novo KIF1A variants, suggesting a possible link between the EEG abnormality and KIF1A variants.10 Our results show that KIF1A variants must be considered in patients with HSP, regardless of the mode of inheritance. The gene REEP2 was also recently associated with both dominant and recessive HSP,24 both inheritance types being classified under SPG72.25 Therefore, we suggest that HSP caused by dominant KIF1A variants be classified in the same group as HSP caused by recessive KIF1A variants, that is, SPG30.
The previous de novo variants clustered in the motor domain of KIF1A between amino acids 58 and 316 and they were predicted to affect different properties of the motor domain function.10 Some substitutions disrupted directly the ATP binding pocket or interfered with the γ-phosphate release through the back door structure after ATP hydrolysis, whereas c.946C>T (p.(Arg316Trp)) affected microtubule binding. The c.206C>T (p.(Ser69Leu)) identified in this study might cause a conformational change that also affects the stable ATP binding but not largely the overall structure of the ATP binding pocket. KIF1A has been suggested to function as a dimer, similarly to the family 1 kinesins KIF5A, KIF5B and KIF5C that move ‘head-over-head’ such that one motor domain (or head) of the dimer binds the microtubule, whereas the other moves over to the next binding site.1, 26, 27 Dominant negative effect is postulated for the heterozygous KIF1A variants,10 which is readily understandable if, in fact, the protein dimerizes.
The two known recessive disease variants of the motor domain6, 7 may have different consequences on the protein’s function than the dominant variants, which could explain why they do not cause disease in heterozygous carriers. Structural modeling suggests that the dominant disease variants affect either ATP binding, γ-phosphate release or microtubule binding.10 On the basis of structural analyses, the recessive variants c.764C>T (p.(Ala255Val)) and c.1048C>G (p.(Arg350Gly)) were predicted to disrupt the back door structure or the neck linker between the motor domain and cargo binding regions, respectively.6, 7, 10 Functional testing by the expression of variants in rat hippocampal neurons suggested that the recessive variants impaired motor activity of KIF1A but to a lesser degree than the dominant disease variants.10 Therefore, a less severe biochemical phenotype may explain why c.764C>T (p.(Ala255Val)) and c.1048C>G (p.(Arg350Gly)) are disease causing only in the homozygous state. Recessive inheritance was also documented for KIF1A variants that cause HSANII.5 These variants are downstream of the motor domain and may therefore not be detrimental to KIF1A motility, but may instead interfere with vesicle binding as the pleckstrin homology domain is disrupted.5 Thus, a relatively less severe biochemical consequence, compared with dominant motor domain variants, may account for heterozygous carriers of the truncating variants being symptom free. Further functional studies are needed to fully elucidate the precise mechanism of genetic dominance and neuron type specificity of KIF1A variants.
In conclusion, our study expands the spectrum of KIF1A-related disease by documenting the first dominantly inherited disease variant. We recommend testing of KIF1A in HSP regardless of the inheritance pattern.
References
Hirokawa N, Nitta R, Okada Y : The mechanisms of kinesin motor motility: lessons from the monomeric motor KIF1A. Nat Rev Mol Cell Biol 2009; 10: 877–884.
Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N : The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 1995; 81: 769–780.
Shin H, Wyszynski M, Huh KH et al: Association of the kinesin motor KIF1A with the multimodular protein liprin-alpha. J Biol Chem 2003; 278: 11393–11401.
Yonekawa Y, Harada A, Okada Y et al: Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol 1998; 141: 431–441.
Riviere JB, Ramalingam S, Lavastre V et al: KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 2011; 89: 219–230.
Erlich Y, Edvardson S, Hodges E et al: Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis. Genome Res 2011; 21: 658–664.
Klebe S, Lossos A, Azzedine H et al: KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur J Hum Genet 2012; 20: 645–649.
Klebe S, Azzedine H, Durr A et al: Autosomal recessive spastic paraplegia (SPG30) with mild ataxia and sensory neuropathy maps to chromosome 2q37.3. Brain 2006; 129: 1456–1462.
Fink JK : Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 2013; 126: 307–328.
Lee JR, Srour M, Kim D et al: De novo Mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy and cerebellar atrophy. Hum Mutat 2014; doi:10.1089/hum.2012.003.
Okamoto N, Miya F, Tsunoda T et al: KIF1A mutation in a patient with progressive neurodegeneration. J Hum Genet 2014; 59: 639–64.
Hamdan FF, Gauthier J, Araki Y et al: Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet 2011; 88: 306–316.
Ylikallio E, Johari M, Konovalova S et al: Targeted next-generation sequencing reveals further genetic heterogeneity in axonal Charcot-Marie-Tooth neuropathy and a mutation in HSPB1. Eur J Hum Genet 2014; 22: 522–527.
Sulonen AM, Ellonen P, Almusa H et al: Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol 2011; 12: R94.
Robinson JT, Thorvaldsdottir H, Winckler W et al: Integrative genomics viewer. Nat Biotechnol 2011; 29: 24–26.
Lim ET, Wurtz P, Havulinna AS et al: Distribution and Medical Impact of Loss-of-Function Variants in the Finnish Founder Population. PLoS Genet 2014; 10: e1004494.
Kumar KR, Blair NF, Vandebona H et al: Targeted next generation sequencing in SPAST-negative hereditary spastic paraplegia. J Neurol 2013; 260: 2516–2522.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Schwarz JM, Cooper DN, Schuelke M, Seelow D : MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014; 11: 361–362.
DeLano W : PyMOL molecular viewer: updates and refinements. Abstr Pap Am Chem Soc 2009; 238: available at www.pymol.org.
Fiser A, Do RK, Sali A : Modeling of loops in protein structures. Protein Sci 2000; 9: 1753–1773.
Kikkawa M, Sablin EP, Okada Y, Yajima H, Fletterick RJ, Hirokawa N : Switch-based mechanism of kinesin motors. Nature 2001; 411: 439–445.
Nitta R, Kikkawa M, Okada Y, Hirokawa N : KIF1A alternately uses two loops to bind microtubules. Science 2004; 305: 678–683.
Esteves T, Durr A, Mundwiller E et al: Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia. Am J Hum Genet 2014; 94: 268–277.
Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A : Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 2014; 261C: 518–539.
Yue Y, Sheng Y, Zhang HN et al: The CC1-FHA dimer is essential for KIF1A-mediated axonal transport of synaptic vesicles in C. elegans. Biochem Biophys Res Commun 2013; 435: 441–446.
Huo L, Yue Y, Ren J et al: The CC1-FHA tandem as a central hub for controlling the dimerization and activation of kinesin-3 KIF1A. Structure 2012; 20: 1550–1561.
Acknowledgements
We would like to thank the family for participation in the study. Riitta Lehtinen is thanked for her technical help. We also acknowledge the target enrichment, sequencing and variant calling pipeline analyses performed by the Institute for Molecular Medicine Finland. We wish to thank the following funding sources for support: Sigrid Jusélius Foundation (for HT), the Finnish Neuromuscular Disorders Association (for MA), University of Helsinki (for HT), the Academy of Finland (for HT and EY), Svenska kulturfonden (for EY), Arvid and Greta Olin’s Foundation (for EY), Finska Läkaresällskapet (for EY) and the Institute for Basic Science (for EK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Ylikallio, E., Kim, D., Isohanni, P. et al. Dominant transmission of de novo KIF1A motor domain variant underlying pure spastic paraplegia. Eur J Hum Genet 23, 1427–1430 (2015). https://doi.org/10.1038/ejhg.2014.297
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.297
This article is cited by
-
Clinical genetics of Charcot–Marie–Tooth disease
Journal of Human Genetics (2023)
-
Monoallelic KIF1A-related disorders: a multicenter cross sectional study and systematic literature review
Journal of Neurology (2022)
-
Challenges and Controversies in the Genetic Diagnosis of Hereditary Spastic Paraplegia
Current Neurology and Neuroscience Reports (2021)
-
KIF1A-related autosomal dominant spastic paraplegias (SPG30) in Russian families
BMC Neurology (2020)
-
KIF1A variants are a frequent cause of autosomal dominant hereditary spastic paraplegia
European Journal of Human Genetics (2020)
