
Abstract
A homozygote missense mutation of the pseudouridylate synthase gene was found in an adult patient with chronic sideroblastic anemia, diarrhea, microcephaly and failure to thrive. Moderate muscle weakness occurred in adulthood (6-min walk distance at 26 years: 240 m, control range 380–782 m) but a profound deficiency of mitochondrial respiratory chain complexes I and IV were found in her skeletal muscle. This, to our knowledge, is the first example of long survival of this usually fatal mitochondrial deficiency into adulthood. We suggest giving consideration to mitochondrial translation deficiency in unexplained syndromic sideroblastic anemia in adulthood.
Similar content being viewed by others


Introduction
Congenital sideroblastic anemia is a rare genetic disease caused by defects of heme biosynthesis and mitochondrial energy production. The most commonly recognized congenital sideroblastic anemias are X-linked sideroblastic anemia due to mutations in the gene encoding delta amino levulinic acid synthase (ALAS2)1 and the Pearson marrow-pancreas syndrome, caused by sporadic, large-scale deletions of mitochondrial DNA.2 Recently, nuclear-encoded disorders of mitochondrial translation have emerged as a novel cause of syndromic sideroblastic anemia with myopathy and lactic acidosis. Indeed, two disease genes affecting mitochondrial translation have been shown to cause syndromic sideroblastic anemia, namely the mitochondrial tyrosyl-tRNA synthetase 2 gene (YARS2)3 and the pseudouridylate synthase gene (PUS1).4
On the other hand, the myopathy, lactic acidosis and sideroblastic anemia syndrome (MLASA) is a rare condition that combines early-onset myopathy with lactic acidosis and sideroblastic anemia. MLASA has been ascribed to mutations in PUS1, an enzyme involved in post transcriptional modification of multiple cytoplasmic and mitochondrial tRNAs (pseudouridylation). We identified a homozygous PUS1 mutation in an adult patient with congenital sideroblastic anemia, diarrhea, microcephaly, short stature, late-onset moderate muscle weakness and a rather favorable clinical outcome at 26 years of age. This report illustrates the unusual expression and occasionally favorable outcome of this condition in adulthood.
Subjects and methods
The patient, the fourth child of healthy first cousin Turkish parents, was born at term after an uncomplicated pregnancy. Three elder sibs are healthy. Aged 1 year, following episodes of pallor and jaundice, she was diagnosed with congenital sideroblastic anemia. Bone marrow aspirate showed dyserythropoiesis and supported the diagnosis of transfusion-dependent congenital sideroblastic anemia (hemoglobin, 6.3 g/l; reticulocytes, 62 000/mm3; white blood cells, 7500/mm3; platelets, 345 000/mm3). She had no muscle, heart or liver involvement, but chronic diarrhea prompted to consider the diagnosis of Pearson marrow-pancreas syndrome.5 Skeletal muscle biopsy at 4 years ruled out mitochondrial DNA deletion, but detected a profound deficiency of mitochondrial respiratory chain complexes I and IV (complex IV/II+III= 0.9, control mean±1 SD: 3.1±0.4; complex IV/III: 0.6, control mean±1 SD: 1.5±0.4). Iterative blood transfusions were well tolerated and she only complained about diarrhea, abdominal pain and occasional headache. She failed to thrive normally; her muscle bulk was reduced but no major muscle weakness, lid ptosis or fatigue was reported. Histological examination of muscle revealed few ragged red fibers (Figure 1a). Neither acute episodes of metabolic acidosis nor neurological symptoms were noted. Plasma lactate consistently remained into normal range. She needed special schooling, but she was an otherwise friendly and outgoing adolescent and young adult. She is now a 26-year-old female, with mild intellectual disability and microcephaly (OFC: 49 cm, −3 SD; weight: 43 kg, −2 SD; height: 153 cm, −1 SD). She can walk unaided with no muscle pain but she recently developed a late-onset, moderate muscle weakness (6-min walk distance at 26 years: 240 m, control range 380–782 m6). She has no cardiac involvement and normal heart ultrasound. Apart for monthly blood transfusions and prevention of hemochromatosis, her major concern is her social and professional future.

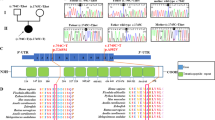
(a) Gomori trichrome staining of muscle biopsy from the patient. Note the presence of few ragged‐red fibers. (b) BN-PAGE analysis of mitochondria from cultured skin fibroblasts of patient (P) and control (C) using specific antibodies. The arrow indicates a putative subcomplex of complex I. (c) Molecular analysis of the PUS1 gene in the patient. The arrows indicate the nucleotide variation. (d) Segregation of the PUS1 mutation in the family. (e) Evolutionary conservation of human Arg294 in PUS1 of various species. Position of the Arg294 is indicated by an arrow.
All samples from the patient were collected in accordance with regulations from the Ethical Committee of the Necker Hospital. Informed consent was given before sample collection.
Blue-Native polyacrylamide gel (BN-PAGE) analysis was carried out on mitoplasts isolated from control and patient cells grown in DMEM GlutaMax media containing 10% FBS, 100 μg/ml streptomycin and 100 U/ml penicillin (Life Technologies, Saint Aubin, France). Cells from a single 75 cm2 flask were grown to ~90% confluence, trypsinized, collected by centrifugation, washed once with PBS and resuspended in 100 μl cold PBS (Life Technologies). Digitonin (4 mg/ml) was added to a final concentration 2 mg/ml. After incubation on ice for 10 min, samples were diluted by addition of 1 ml cold PBS. Mitoplasts were recovered by a centrifugation at 16 000 g for 10 min at 4oC and resuspended in 30 μl ACBT buffer (1.5 M aminocaproic acid, 75 mm Bis-Tris, pH 7.0) containing 1% w/v N-Dodecyl-β-d-maltoside. Protein concentration was measured using Bradford reagent (BioRad, Marnes-la-Coquette, France) and 20 μg of mitoplast protein were fractionated through a 4–16% NativePage gel (Life Technologies) according to manufacturer’s recommendations. Finally, OXPHOS complexes were electro blotted on PVDF membranes (GE Healthcare, Aulnay sous Bois, France) and detected by immunostaining with antibodies directed against proteins from individual OXPHOS complexes (Abcam, Cambridge, UK): anti-GRIM19 for complex I (CI), anti-SDHA for complex II (CII), anti -core II for complex III (CIII), anti-COXII for complex IV (CIV) and anti-F1β subunits for complex V (CV).
Patient's genomic DNA (1 μg) was isolated from blood leukocytes. Exons were captured by an in-solution enrichment methodology (SureSelect Human All Exon Kits Version 3, Agilent, Les Ulis, France) using a biotinylated oligonucleotide probe library (Human All Exon v3 50 Mb, Agilent).
Gomori trichome staining was performed as previously described.7
Results
BN-PAGE on cultured skin fibroblasts revealed qualitative anomalies in assembly of complex I respiratory enzyme (Figure 1b), reminiscent of anomalies observed in fibroblasts of patients with mitochondrial translation deficiencies.8,9
To identify the causative nuclear gene mutation, we sequenced the exome of this patient. From the 22 697 SNPs and Indels identified, the pathogenic variant was selected after filtering against known SNPs reported in dbSNP, 1000 genomes, Exome Variant Server, Intergenic variants and in house polymorphisms. Considering the consanguinity of the parents, only homozygous variations were considered. This filtering resulted in a list of 14 genes, only one of which encoding a known mitochondrial protein, PUS1. Sanger sequencing confirmed this homozygous missense mutation in exon 5 of the PUS1 gene (c.883 C>T, p.Arg295Trp, Figure 1c). The parents were heterozygous for this mutation affecting a conserved codon (Figures 1d and e), which was predicted to be deleterious by various prediction softwares (SIFT: deleterious (score: 0.00, median: 3.02); MutationTaster: disease causing (P-value: 1.0); Polyphen2: probably damaging, score 1.00010,11).
Discussion
PUS1 mutations have been rarely reported. Systematic analysis of PUS1 in a large series of 60 patients with sideroblastic anemia allowed to exclude this gene in all patients but two who had MLASA.12 Moreover, PUS1 mutations have also been excluded as a cause of acquired sideroblastic anemia in a series of 37 patients.13 Until now, all patients with PUS1 mutations have MLASA.4,12,14,15 Our rather unusual observation emphasizes the clinical variability of the disease and the occasionally long survival of congenital mitochondrial translation deficiency. The variable expressivity and relatively favorable outcome of this case of PUS1 mutation, compared with the severity of most reported cases, remains unexplained. Trying to elucidate the underlying mechanisms of this favorable outcome should certainly help improving the long-term prognosis of respiratory chain deficiency into adulthood. PUS1 encodes a mitochondrial tRNA modification factor and mutations of this gene have been showed to result in abnormal mitochondrial translation.14 Mitochondrial translation deficiencies often result in quantitative and/or qualitative respiratory chain assembly defects in fibroblasts but this feature has never been described for PUS1 mutations to our knowledge. Therefore, we suggest to test PUS1 in patients with isolated or syndromic sideroblastic anemia associated with abnormal respiratory chain assembly pattern.
References
Cotter PD, Rucknagel DL, Bishop DF : X-linked sideroblastic anemia: identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood 1994; 84: 3915–3924.
Rotig A, Colonna M, Bonnefont JP et al. Mitochondrial DNA deletion in Pearson's marrow/pancreas syndrome. Lancet 1989; 1: 902–903.
Riley LG, Cooper S, Hickey P et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am J Hum Genet 2010; 87: 52–59.
Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N : Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet 2004; 74: 1303–1308.
Bader-Meunier B, Rotig A, Mielot F et al. Refractory anaemia and mitochondrial cytopathy in childhood. Br J Haematol 1994; 87: 381–385.
Casanova C, Celli BR, Barria P et al. The 6-min walk distance in healthy subjects: reference standards from seven countries. Eur Respir J 2011; 37: 150–156.
Gomori G : A rapid one-step trichrome stain. Am J Clin Pathol 1950; 20: 661–664.
Galmiche L, Serre V, Beinat M et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat 2011; 32: 1225–1231.
Vedrenne V, Galmiche L, Chretien D, de Lonlay P, Munnich A, Rotig A : Mutation in the mitochondrial translation elongation factor EFTs results in severe infantile liver failure. J Hepatol 2012; 56: 294–297.
Kumar P, Henikoff S, Ng PC : Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
Adzhubei IA, Schmidt S, Peshkin L et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Bergmann AK, Campagna DR, McLoughlin EM et al. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatric blood & cancer 2010; 54: 273–278.
Steensma DP, Hecksel KA, Porcher JC, Lasho TL : Candidate gene mutation analysis in idiopathic acquired sideroblastic anemia (refractory anemia with ringed sideroblasts). Leuk Res 2007; 31: 623–628.
Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, Zeviani M : Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J Med Genet 2007; 44: 173–180.
Zeharia A, Fischel-Ghodsian N, Casas K et al. Mitochondrial myopathy, sideroblastic anemia, and lactic acidosis: an autosomal recessive syndrome in Persian Jews caused by a mutation in the PUS1 gene. J Child Neurol 2005; 20: 449–452.
Acknowledgements
This work was supported by INSERM and Association Française contre les Myopathies (AFM). MDM is supported by a fellowship from the AFM (16615).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Metodiev, M., Assouline, Z., Landrieu, P. et al. Unusual clinical expression and long survival of a pseudouridylate synthase (PUS1) mutation into adulthood. Eur J Hum Genet 23, 880–882 (2015). https://doi.org/10.1038/ejhg.2014.192
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.192
