
Abstract
This article describes the inter- and intra-familial phenotypic variability in four families with WNT10A mutations. Clinical characteristics of the patients range from mild to severe isolated tooth agenesis, over mild symptoms of ectodermal dysplasia, to more severe syndromic forms like odonto-onycho-dermal dysplasia (OODD) and Schöpf–Schulz–Passarge syndrome (SSPS). Recurrent WNT10A mutations were identified in all affected family members and the associated symptoms are presented with emphasis on the dentofacial phenotypes obtained with inter alia three-dimensional facial stereophotogrammetry. A comprehensive overview of the literature regarding WNT10A mutations, associated conditions and developmental defects is presented. We conclude that OODD and SSPS should be considered as variable expressions of the same WNT10A genotype. In all affected individuals, a dished-in facial appearance was observed which might be helpful in the clinical setting as a clue to the underlying genetic etiology.
Similar content being viewed by others


Introduction
Tooth agenesis (TA) is the most common, often heritable, developmental anomaly of the human dentition. Depending on the tooth type and number of teeth involved, the prevalence of TA ranges from 10 to 30% for TA of third molars to 0.2% for severe TA of six teeth or more (excluding the third molars).1 An overall incidence of TA of permanent teeth excluding third molars of 2.6–11.3% was reported, depending on demographic and geographic profiles.1
As tooth formation shares mechanisms with the development of other ectodermal appendages, TA often occurs as part of a variety of ectodermal dysplasia (ED) syndromes including Witkop syndrome, hypohydrotic ectodermal dysplasia, odonto-onycho-dermal dysplasia (OODD), Schöpf–Schulz–Passarge syndrome (SSPS) and others. For these human TA syndromes, mutations have been identified, respectively, in MSX1,2 EDA, EDAR,3, 4, 5 EDARADD,6 and WNT10A.7, 8 WNT10A mutations were identified in a family of which four members were affected with isolated TA or microdontia,9 whereas more recently in a larger cohort of 34 patients with isolated TA, 56% appeared to have WNT10A mutations.10
Besides pathogenic mutations within the coding regions of the WNT10A gene in 62% of the tested patients, a significant association between SNPs in the WNT10A gene and the risk for TA was found.11 These recent studies indicate that changes in WNT10A are highly frequent in terms of the risk or the presence of isolated or syndromic hypodontia, presenting with variable phenotypes and genotypes, including homozygous, compound heterozygous and heterozygous mutations.
It has been shown that specific facial features can differ significantly between patients with and without isolated TA.12 Therefore, in addition to the description of the dental and general syndromic ED features, the aim in this study was to document the facial phenotypic variations in four families with WNT10A mutations and to give a comprehensive overview of the literature regarding WNT10A mutations, associated conditions and developmental defects.
Patients and methods
In four families at least one member, referred to as proband or index patient, was referred for diagnosis or treatment to the Department of Orthodontics and Craniofacial Biology, the Radboud Center for Cleft Palate and Craniofacial Anomalies and/or the Center for Special Dental Care or the Department of Human Genetics, Radboud University Medical Center (RUMC). Informed consent for publication of data and photographs was obtained from all patients and their enrolled relatives.
Patients referred for orthodontic treatment received full diagnostic records, including digital or plaster dental casts, a lateral headfilm and orthopantomogram (OPT) (In order to allow an exact quantitative phenotype profiling, the term TA with addition of the exact number of teeth which failed to develop (excluding the third molars) is used as the terms hypodontia and oligodontia can be misleading.13 To identify the present/absent teeth, the Fédération Dentaire Internationale tooth numbering system is used.). Both intra- and extra-oral two-dimensional (2D) photographs as well as facial 3D stereophotogrammetry images were made. The 3D surface images were taken with a 3dMDface system (3dMD, Atlanta, GA, USA). All data were separately evaluated by two staff orthodontists of the Department of Orthodontics and Craniofacial Biology of the RUMC.
A medical and dental history was taken, including the family history; a full physical examination was also performed by a clinical geneticist. The patients’ DNA was isolated from leukocytes from their peripheral blood samples according to a standard protocol. Sequence analysis of WNT10A (RefSeq: NM_025216.2) was performed at the DNA diagnostics laboratory of the University Medical Center Utrecht, The Netherlands, using the standard Sanger sequencing technique. After identifying the mutations in the probands their first degree relatives were contacted and were asked to participate in the study.
The clinical examination and blood samples of the enrolled family members were taken in the same way as in the index patient while the oral and facial documentation in the family members was limited to a clinical examination and 2D facial photographs.
Complete documentation was obtained with regard to all index patients except one (patient D II-1). At the time of inclusion this patient was already treated prosthodontically and no adequate documentation could be retrieved from the period before treatment. Also for some of the first degree relatives included in this study, only limited data were available.
Results
Overview literature regarding WNT10A mutations
On the 13th of March 2013, a literature search was performed, screening the NCBI database for WNT10A mutations. Filters only allowing articles in English, German or Dutch, yielded 90 articles. When adding Tooth Agenesis AND Ectodermal Dysplasia, Odonto-Onycho-Dermal Dysplasia, Schöpf–Schulz–Passarge Syndrome AND/OR derivates to WNT10A mutations, 14 articles7, 8, 9, 10, 11, 14, 15, 16, 17, 18, 19, 20, 21, 22 met the inclusion criteria and suggested a causative relationship of WNT10A mutations with isolated TA or/and ED phenotypes.
Adaimy et al14 were the first authors to relate a homozygous mutation in the WNT10A gene to OODD, while Bohring et al15 were the first to describe SSPS in patients with WNT10A mutations.
To date, 28 different mutations resulting in 39 different compound heterozygous, heterozygous or homozygous WNT10A mutations, leading to TA with or without ED symptoms, have been reported in 139 patients. In the homozygous/heterozygous patients, one frameshift mutation occurred, one nonsense mutation and eight different missense mutations were detected. In the compound heterozygous patients, 1 missense/frameshift mutation, 4 nonsense/nonsense, 8 nonsense/missense mutations and 16 different missense/missense mutations are identified. In the group of nonsense mutations, p.(Cys107*) is the most common, and the p.(Phe228Ile) mutation is most frequent in the missense mutations. The articles with the identified mutations and associated phenotypes are listed alphabetically in Table 1 and indicated/displayed schematically in Figure 1.

Chromosome 2 with location and spread of reported mutations within the WNT10A gene. Schematic representation of the location of the WNT10A gene with its four exons within chromosome 2. Displayed nucleotide/amino-acid substitutions correspond with the currently known WNT10A mutations described in Table 1. Boxed substitutions are the mutations identified in this study.
Description of four families with WNT10A mutations
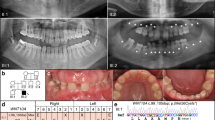
An overview of the WNT10A mutations, phenotypic characteristics and clinical diagnosis of the four families examined in this study is also displayed in Table 2 and Figure 2a–d.

a–d: Genotype and clinical and orofacial phenotype of four WNT10A mutation families. Pedigree as well as documentation from the probands (2D facial photographs, 3D digital stereophotogrammetry, intra-oral photographs and OPT) is displayed per family. Documentation from index patients in family A–C dates from the period prior to any orthodontic, surgical and/or comprehensive prosthetic treatment. Only patient D II-1 already had comprehensive prosthodontic treatment and adequate pre-treatment documentation could not be retrieved. Probands from family A–C consented to publication of all documentation, Patient D II-1 consented only to publication of intra-oral photographs and OPT.
Family A
Clinical examination proband. The proband is a 9-year-old boy of Belgian descent (patient A II-2) with TA of 22 permanent teeth. Only six permanent teeth (16, 11, 21, 26, 36 and 46) developed while his mother reported that her son had a complete deciduous dentition. The upper permanent central incisors are conically shaped and the upper first permanent molars are hypoplastic. There is spacing between the front teeth, and the maxillary central incisors are mildly retroclined. The mandibular dental arch has a more dorsal position relative to the maxillary dental arch (Class II/1 relation). The 3D facial image demonstrates an underlying hypodivergent skeletal pattern, with a retrusive mandible and a deepened plica mentalis.
Additional ED features are present, comprising nail dystrophy and keratodermata at his feet. Temporary alopecia of the skull hair had occurred at the age of 7. He was clinically diagnosed with OODD.
Clinical examination of the first degree relatives. Both the proband’s father and his brother show no clinical symptoms of ED and all teeth are present. His mother however misses one tooth (35), and shows generalized small-sized teeth with various diastemas in both jaws. Like her youngest son, she showed some retroclination of her maxillary front teeth, prior to her orthodontic treatment.
Genetic diagnosis. A compound heterozygous mutation was identified in WNT10A: c.321C>A;682T>A, p.(Cys107*);(Phe228Ile). In either parent the presence of a heterozygous change in WNT10A was confirmed, the asymptomatic father harbored the c.321C>A, p.(Cys107*) mutation and the c.682T>A, p.(Phe228Ile) mutation was identified in the mother who was missing one tooth. The mutation analysis in the proband’s brother revealed he is a heterozygous carrier of the c.321C>A, p.(Cys107*) mutation, which was inherited from the father (see Table 2).
Family B
This Dutch family was referred to the Department of Orthodontics and Craniofacial Biology because of TA in their three children. In all family members, the deciduous dentition was either not affected or could not be verified.
Clinical examination proband. The proband was 19 years old at the time of inclusion (patient B II-3) with severe TA missing 12 permanent teeth (17, 13, 11, 21, 23, 24, 27, 32, 33, 34, 35, 37, 42, 43, 44, 47). The upper anterior teeth show a conical aspect and the permanent upper first molars are hypoplastic. Owing to a transverse maxillary hypoplasia, a bilateral cross-bite is present in the molar region (including permanent and deciduous molars). In contrast to the prominent chin point at pogonion, the 3D facial image indicates the underlying basal bone and its alveolar ridge to be retrusive. The deep mental fold and the flattened lip profile, give rise to a dished-in profile.
His hair is thin, grows slowly and he suffered from frontal balding at a young age. However, his body hair as well as his nails grow normally. He suffered from eczema during childhood. At age 7, he suffered from avascular bone necrosis of the os naviculare (Morbus Köhler). At the time of examination, he reported to have recurrent joint pains, diagnosed as fibromyalgia. Other clinical observations are the multiple skin naevi and clinodactyly of the fifth digits of both hands. Based on the above findings and the family history, the proband was diagnosed with SSPS.
Clinical examination of the first degree relatives. The oldest sister of the proband was 23 years old (patient B II-1) when examined. Her mother reported her birth had been uneventful, but in the neonatal period she was admitted to the hospital because of sudden collapses. No cause for these collapses had been identified and these events were self-limiting. Like her brother and sister she also suffers from joint pains, but no dysmorphic oral features are recorded. Physical examination shows a young woman with fair, normally growing hair but with thin and brittle nails. She has multiple skin naevi, two of which had been removed recently. One of them was reported to be difficult to diagnose pathologically and was classified as a high-graded Spitzoïd Tumor of Uncertain Malignant Potential or Spitzoïd type Malignant Melanoma. The other was classified as a regular naevus naevocellularis.
Intra-oral examination showed TA of nine permanent teeth (17, 12, 22, 25, 27, 37, 35, 41 and 47), excluding the third molars. Like her brother she also has a narrow maxilla. Regardless the normal extra-oral proportions of her face, she also shows tendency toward a dished-in facial profile. Like her brother she was diagnosed with SSPS.
The probands’ second sister (patient B II-2) was 21 years old when first seen and showed TA of only the 2 upper lateral incisors. As a child, she suffered from eczema. Nowadays her hair and nails grow normally. She reported to have experienced recurrent painful swellings of one eyelid. She has joint pains which were diagnosed as fibromyalgia, but an autoimmune disease could not be excluded. On physical examination, several café-au-lait macules were seen and multiple skin naevi were also present, mostly on her back. The facial features were again a slightly retrusive mandible as well as a dished-in profile.
The children’s mother reported to be healthy, but she also suffers from joint pains. She has a complete permanent dentition and shows no signs or symptoms of ED. The father has a complete permanent dentition as well. His hair grows normally and he has no skin abnormalities, however he shows brittle nails. He suffers from amblyopia.
Genetic diagnosis. A compound heterozygous mutation in WNT10A was identified in this proband (B II-3): c.321C>A;682T>A, p.(Cys107*);(Phe228Ile). The oldest sister (B II-2) also shows the same compound heterozygous mutation as the proband: c.321C>A;682T>A, p.(Cys107*);(Phe228Ile). The youngest sister (B II-1) is a heterozygous carrier of the c.682T>A, p.(Phe228Ile) mutation which she inherited from her mother. Father and mother are heterozygous carriers of, respectively, the c.321C>A, p.(Cys107*) and c.682T>A, p.(Phe228Ile) mutation.
Family C
The proband (patient C II-1) was seen in the Clinical Genetics Department at age 19. She was born to Dutch consanguineous parents.
Clinical examination proband. As a baby she was bald for a long time, and currently her hair growth is still impaired for which she is wearing hair extensions. Her eyebrows are sparse and she shows brittle nails. She has dry skin and sweating is impaired as well.
She suffers from severe TA of 19 permanent teeth, only 15, 14, 11, 21, 23, 35, 34, 44 and 45 developed. The tongue had a smooth surface probably caused by a reduced number of papillae. In addition, she shows a hypoplastic maxilla in the transverse as well as sagittal directions resulting in bilateral cross-bites in the premolar area. Again the dished-in profile is a prominent facial feature which is more pronounced due to a protruded chin point at pogonion.
Clinical examination of the first degree relatives. The mother of the proband has mild TA of two teeth (12 and 14). She reported her scalp hair and eyebrows growing slowly. She has difficulty sweating and has brittle nails. The father of the proband has a complete permanent dentition. He shows dry skin and somewhat brittle nails (which may be due to nail biting) but no other signs of ED are recorded.
Genetic diagnosis. The proband shows a homozygous nonsense WNT10A mutation: c.321C>A;321C>A, p.(Cys107*); (Cys107*). Both parents are heterozygous carriers of the c.321C>A mutation.
Family D
The proband (patient D II-2) is a 24-year-old Dutch woman referred to the Clinical Genetics Department because of ED symptoms.
Clinical examination proband. In her permanent dentition, she has severe TA of 23 teeth (17, 15, 14, 12, 11, 21, 22, 24, 25, 26, 27, 37, 36, 35, 34, 32, 31, 41, 42, 43, 44, 45 and 47), leaving her with only five permanent teeth present (16, 13, 23, 33 and 46), two of which have a conical shape and one being hypoplastic. Although her mandible is also retruded, leading to a convex profile, the dished-in appearance is again the most catching feature in her facial profile.
Moreover she has sparse, thin hair and brittle nails which grow slowly. She has difficulty sweating in high temperatures and has myopia of −4 and −2.
Clinical examination of the first degree relatives. The proband has two brothers. One brother (D II-1) has mild TA with only one tooth missing and a one hypoplastic molar and the other (D II-3) shows a complete permanent dentition with normal morphology of the teeth. In both brothers no signs of ED are present.
The probands’ mother has mild TA with two teeth missing. She also shows brittle nails and suffers from glaucoma. In the mother’s family, there were several individuals with mild ED symptoms, like conically shaped teeth and mild TA. The father misses one tooth.
Genetic diagnosis. The proband shows a homozygous nonsense WNT10A mutation: c.321C>A;321C>A, p.(Cys107*);(Cys107*). Both parents are heterozygous carriers of the c.321C>A mutation.
Discussion
For this study clinical and molecular data of 15 family members from four unrelated families diagnosed with WNT10A mutations were collected and documented. Homozygous, heterozygous as well as compound heterozygous WNT10A mutations were identified (Table 2). These patients were affected with variable TA phenotypes with or without syndromic ED features and with overlapping OODD and SSPS syndromes.
The most severe TA phenotype (23 permanent teeth failed to develop) was found in the female proband of family D (D II-1) in whom TA was associated with mild ED symptoms (Table 3). This patient carried a homozygous nonsense WNT10A mutation c.321C>A;321C>A, p.(Cys107*);(Cys107*) causing a premature stop codon and thus a truncated protein at p.(Cys107*) (Table 2) which might also be accompanied by nonsense-mediated decay of the (pre) mRNA. In Family C the female II-1 proband carrying the same homozygous c.321C>A nonsense mutation shows a milder TA phenotype (with 19 teeth missing) and an even larger difference in TA phenotype (22 vs 12 teeth missing) exists between the two probands of Family A and B, both carriers of the same compound heterozygous nonsense/missense mutation c.321C>A;682T>A, p.(Cys107*);(Phe228Ile). Moreover, their associated ED phenotypes pointed to the different clinical entities of OODD and SSPS, respectively, which refutes the genotype–phenotype correlation hypothesis for mutations within the WNT10A gene.
In our cohort of families with WNT10A mutations, we found that the most frequently observed mutations in the literature, c.321C>A;682T>A, p.(Cys107*);(Phe228Ile) were present in all affected members in heterozygous, compound heterozygous or homozygous form (Table 2). Although recorded in different configurations, these gene variations caused the expression of a variety of phenotypes in the four patients with a mutation on both alleles.
Bohring et al15 stated that ∼50% of obligate heterozygotes for WNT10A mutation display clinical symptoms of ED. In our four families, the relatives of the patients who were all confirmed heterozygous carriers show very mild or no distinctive phenotype at all, except for the mother in family C (patient I-3) carrying a heterozygous c.321C>A, p.(Cys107*) mutation who shows several distinct characteristics of ED. These findings demonstrate the variety in penetrance of symptoms in patients and the probability of other currently unknown influences (genetic, epigenetic or environmental) that may be relevant to WNT signaling and to ectodermal homeostasis as suggested in recent studies.8, 18, 19, 20
Recently a double entrance of c.682T>A, p.(Phe228Ile) in a pedigree was recorded and it was suggested that this specific mutation may be a common finding in the general population.19 In 1 of 400 individuals, a c.682T>A, p.(Phe228Ile) mutation can be seen and in 50% of these cases this mutation would result in developmental ectodermal defects. Foregoing considered, one might question whether OODD and SSPS are actually different disorders. Wedgeworth et al19 noticed eyelid cysts that resolved spontaneously before clinical assessment was performed and concludes that this particular feature should perhaps be regarded as just one physical sign in the OODD/SSPS spectrum rather than a pathognomonic hallmark of SSPS. This conclusion as well as the weak genotype–phenotype correlation mentioned earlier strengthens the assumption that OODD and SSPS are variable expressions of the same syndrome. Other coincident factors, like the individual genomic background or other factors that influence differential gene expression, might cause the phenotypic variation. However, larger studies with accurate patient history should be performed to confirm these assumptions.
It should be noted, that these eyelid cysts have often been reported in association with several skin malignancies.15, 19, 22 Therefore, when OODD is considered as a not fully expressed form of SSPS, OODD/SSPS patients may have an increased risk to develop skin malignancies. This is strengthened by the diagnosis of a high-graded Spitzoïd tumor of uncertain malignant potential or Spitzoïd-type malignant melanoma in patient B II-1. Though the correlation between WNT10A mutations and increased risk for malignancies has never been proven, we would advise patients who are diagnosed with OODD/SSPS to be observant on the development of skin anomalies and to undergo routine dermatological surveillance. Combined with genetic counseling this also can be helpful in identifying high risk patients.
Besides pathogenic mutations within the coding region of the WNT10A gene, a significant association between SNPs in the WNT10A gene and the risk for TA was found.11 Moreover, the study showed borderline association between the risk of non-syndromic TA and APC, CTNNB1, DVL2 and WNT11 polymorphisms. The authors concluded that variants of genes encoding important components of the WNT signaling pathway might influence the risk of TA. Lammi et al23 already showed the association of TA and increased risk of colorectal cancer for AXIN2, another gene from the WNT signaling pathway. Their results provided the first evidence of the importance of WNT signaling for the development of the dentition in humans and suggest that an intricate control of WNT signal activity is necessary for normal tooth development, since both inhibition and stimulation of WNT signaling due to loss or gain of function mutations in AXIN2 may lead to TA.
In our study, we also confirmed earlier observations that the permanent teeth which did develop in patients with severe TA associated with WNT10A mutations also show various abnormal morphological features of hypoplasia, including conical shapes and microdontia.9, 24 Moreover, the predisposition of ‘second teeth’ agenesis pattern – upper second incisors, upper and lower second premolars and upper and lower second molars – is present in most patients which is in accordance with the findings described by Polder et al in 2004.1
Profile analysis was performed on both intra- and extra oral 2D photographs as well as facial 3D stereophotogrammetric images. Looking at the common 3D facial features we noticed that all affected individuals with severe TA in these four unrelated families show a dished-in maxillofacial appearance and a mildly retruded mandible, pointing to decreased jaw and alveolar bone growth. Intra-orally this alveolar hypoplasia often leads to a uni- or bilateral cross-bite. Despite the mandibular retrusion, the chinpoint at pogonion was prominent in all index patients. This could be the result of decreased posterior occlusal support which also causes a reduced lower anterior face height as seen in most patients.
Hypoplasia of the facial skeleton, especially of the bony alveolar processes of the maxilla and the mandible, when not harboring teeth, is a frequent observation in clinical orthodontics and implantology when treating patients with multiple TA. So far it was accepted that the presence of the teeth themselves within the alveolar processes mechanically models the volume of the jaws.12, 25 Until recently, data suggestive for a possible molecular underpinning of this phenomenon was however lacking. In their study in mice, Cawthorn et al26 identified Wnt10a as well as Wnt10b and Wnt6, as potent endogenous regulators of mesenchymal stem cell fate, inhibiting adipogenesis and stimulating osteoblastogenesis through a β-catenin-dependent mechanism. We therefore hypothesize that mutant WNT10A ligand proteins not being able to initiate the appropriate signaling in the WNT signaling pathway, not only fail to initiate tooth development, but also disrupt the process of osteoblastogenesis ultimately leading to reduced bone mass formation. This could represent the molecular link between TA and reduced osteogenesis, also in facial bone formation, which implicates the hypoplasia of the alveolar processes of the maxilla and the mandible. The fact that all patients with homozygous or compound heterozygous WNT10A mutations showed hypoplasia of their facial skeleton in both transverse and sagittal directions, resulting in an overall dished-in profile, could be indicative for impaired facial bone growth. This is in accordance with the cephalometric finding of Acharya et al12 that severe TA is associated with reduced jaw growth especially of the maxilla.
The above effects attributed to mutations in the WNT10A gene can be explained by its function as a member of the WNT gene family. WNT genes are involved in hair-follicle morphogenesis,27, 28 skin and tooth embryogenesis29, 30, 31 and several other developmental processes.32 It also explains that in addition to TA, hypoplastic morphological dental features, including conical tooth shapes and microdontia, occur in the permanent teeth which did develop.
To our knowledge, this is the first article focusing on the comprehensive description of dental, oral and facial characteristics related to mutations in the WNT10A gene. Early diagnosis is essential in view of a multidisciplinary approach in which also follow-up, regarding the possible risk for the development of skin neoplasms, should be considered. The dentist might be the first and designated person to refer a suspected patient for genetic counseling. However, since the phenotypic spectrum of WNT10A mutations is extremely variable, and the deciduous dentition often shows no abnormalities, one should be aware of other clinical signs of ED and also the facial features like a dished-in appearance.
It is concluded that the clinical phenotypes caused by WNT10A mutations, including isolated TA, with or without ED features and syndromic forms such as OODD and SSPS, vary considerably among individuals and among family members with identical WNT10A mutations. OODD and SSPS should be seen as variable expressions of the WNT10A mutation spectrum instead of different clinical entities.
We observed the presence of a typical facial dished-in appearance in all individuals affected with WNT10A mutations compared with the average norm of facial appearance of non-affected individuals. We hypothesize that, contrary to the TA phenotype and other ED symptoms, this typical facial dished-in aspect could be helpful in the clinical setting to discriminate between the different underlying genetic etiological factors causing the TA phenotype. This however remains to be confirmed in a cohort with TA phenotypes of different known genetic etiologies.
Accession codes
References
Polder BJ, Van 't Hof MA, Van der Linden FP, Kuijpers-Jagtman AM : A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent Oral Epidemiol 2004; 32: 217–226.
Jumlongras D, Bei M, Stimson JM et al: A nonsense mutation in MSX1 causes Witkop syndrome. Am J Hum Genet 2001; 69: 67–74.
Kere J, Srivastava AK, Montonen O et al: X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 1996; 13: 409–416.
Elomaa O, Pulkkinen K, Hannelius U, Mikkola M, Saarialho-Kere U, Kere J : Ectodysplasin is released by proteolytic shedding and binds to the EDAR protein. Hum Mol Genet 2001; 10: 953–962.
Naqvi SK, Wasif N, Javaid H, Ahmad W : Two novel mutations in the gene EDAR causing autosomal recessive hypohidrotic ectodermal dysplasia. Orthod Craniofac Res 2011; 14: 156–159.
Bergendal B, Klar J, Stecksén-Blicks C, Norderyd J, Dahl N : Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Genet A 2011; 155A: 1616–1622.
Nawaz S, Klar J, Wajid M et al: WNT10A missense mutation associated with a complete odonto-onycho-dermal dysplasia syndrome. Eur J Hum Genet 2009; 17: 1600–1605.
Nagy N, Wedgeworth E, Hamada T, White JM, Hashimoto T, McGrath JA : Schöpf-Schulz-Passarge syndrome resulting from a homozygous nonsense mutation in WNT10A. J Dermatol Sci 2010; 58: 220–222.
Kantaputra P, Sripathomsawat W : WNT10A and isolated hypodontia. Am J Med Genet Part A 2011; 155A: 1119–1122.
van den Boogaard MJ, Créton M, Bronkhorst Y et al: Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet 2012; 49: 327–331.
Mostowska A, Biedziak B, Zadurska M, Dunin-Wilczynska I, Lianeri M, Jagodzinski P : Nucleotide variants of genes encoding components of the WNT signalling pathway and the risk of non-syndromic tooth agenesis. Clin Genet 2012; 84: 429–440.
Acharya PN, Jones SP, Moles D, Gill D, Hunt NP : A cephalometric study to investigate the skeletal relationships in patients with increasing severity of hypodontia. Angle Orthod 2010; 80: 699–706.
Nieminen P : Genetic basis of tooth agenesis. J Exp Zool B Mol Dev Evol Part B 2009; 312B: 320–342.
Adaimy L, Chouery E, Megarbane H et al: Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet 2007; 81: 821–828.
Bohring A, Stamm T, Spaich C et al: WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet 2009; 85: 97–105.
van Geel M, Gattas M, Kesler Y et al: Phenotypic variability associated with WNT10A nonsense mutations. Br J Dermatol 2010; 162: 1403–1406.
Cluzeau C, Hadj-Rabia S, Jambou M et al: Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic anhidrotic ectodermal dysplasia cases. Hum Mutat 2011; 32: 70–72.
Castori M, Castiglia D, Brancati F et al: Two families confirm Schöpf-Schulz-Passarge syndrome as a discrete entity within the WNT10A phenotypic spectrum. Clin Genet 2011; 79: 92–95.
Wedgeworth EK, Nagy N, White JM, Pembroke AC, McGrath JA : Intra-familial variability of ectodermal defects associated with WNT10A mutations. Acta Derm Venereol 2011; 91: 346–347.
Petrof G, Fong K, Lai-Cheong JE, Cockayne SE, McGrath JA : Schöpf-Schulz-Passarge syndrome resulting from a homozygous nonsense mutation, p.Cys107*, in WNT10A. Australas J Dermatol 2011; 52: 224–226.
Plaisancié J, Bailleul-Forestier I, Gaston V et al: Mutations in WNT10A are frequently involved in oligodontia associated with minor signs of ectodermal dysplasia. Am J Med Genet A 2013; 161A (4): 671–678.
Starink TM : Eccrine syringofibroadenoma: multiple lesions representing a new cutaneous marker of the Schöpf syndrome, and solitary nonhereditary tumors. J Am Acad Dermatol 1997; 36: 569–576.
Lammi L, Arte S, Somer M et al: Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 2004; 74: 1043–1050.
De Coster PJ, Marks LA, Martens LC, Huysseune A : Dental agenesis: genetic and clinical perspectives. Review. J Oral Pathol Med 2009; 38: 1–17.
van der Linden FPGM, Duterloo HS : Development of the Human Dentition. An Atlas. Harper & Row: Hagerstown, Maryland, 1976.
Cawthorn WP, Bree AJ, Yao Y et al: Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 2012; 50: 477–489.
Andl T, Reddy ST, Gaddapara T, Millar SE : WNT signals are required for the initiation of hair follicle development. Dev Cell 2002; 2: 643–653.
Zhang Y, Tomann P, Andl T et al: Reciprocal requirements for EDA/EDAR/NF-kappaβ and WNT/beta-catenin signaling pathways in hair follicle induction. Dev Cell 2009; 17: 49–61.
Garriock RJ, Warkman AS, Meadows SM, D'Agostino S, Krieg PA : Census of vertebrate WNT genes: isolation and developmental expression of Xenopus WNT2, WNT3, WNT9a, WNT9b, WNT10A, and WNT16. Dev Dyn 2007; 236: 1249–1258.
Kratochwil K, Galceran J, Tontsch S, Roth W, Grosschedl R : FGF4, a direct target of LEF1 and WNT signaling, can rescue the arrest of tooth organogenesis in Lef1(-/-) mice. Genes Dev 2002; 16: 3173–3185.
Yamashiro T, Zheng L, Shitaku Y et al: WNT10A regulates dentin sialophosphoprotein mRNA expression and possibly links odontoblast differentiation and tooth morphogenesis. Differentiation 2007; 75: 452–462.
O'Neill MFJ 2012 *606268 Wingless-type MMTV integration site family, member 10a; WNT10Ahttp://www.omim.org/entry/606268?search=wnt10a&highlight=wnt10a Accessed by the first author on the 13th of March 2013..
Acknowledgements
First of all, we are very grateful to the patients and their families for their willingness to cooperate. The authors also wish to thank the Drs M Van Vlierberghe, Drs R Noverraz and Drs R Pikaar for referral and the provision of documentation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Vink, C., Ockeloen, C., ten Kate, S. et al. Variability in dentofacial phenotypes in four families with WNT10A mutations. Eur J Hum Genet 22, 1063–1070 (2014). https://doi.org/10.1038/ejhg.2013.300
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.300
Keywords
This article is cited by
-
Prevalence of WNT10A gene mutations in non-syndromic oligodontia
Clinical Oral Investigations (2019)
-
Abnormal primary and permanent dentitions with ectodermal symptoms predict WNT10A deficiency
BMC Medical Genetics (2016)
-
Odonto-onycho-dermal dysplasia in a patient homozygous for a WNT10A nonsense mutation and mild manifestations of ectodermal dysplasia in carriers of the mutation
BMC Dermatology (2016)
-
Tooth agenesis and orofacial clefting: genetic brothers in arms?
Human Genetics (2016)
-
Hautveränderungen an Händen und Füßen
Der Hautarzt (2014)
