
Abstract
Warburg Micro Syndrome is a rare, autosomal recessive syndrome characterized by microcephaly, microphthalmia, microcornia, congenital cataracts, optic atrophy, cortical dysplasia, in particular corpus callosum hypoplasia, severe mental retardation, spastic diplegia, and hypogonadism. We have found five new mutations in the RAB3GAP1 gene in seven patients with suspected Micro Syndrome from families with Turkish, Palestinian, Danish, and Guatemalan backgrounds. A thorough clinical investigation of the patients has allowed the delineation of symptoms that are consistently present in the patients and may aid the differential diagnosis of Micro Syndrome for patients in the future. All patients had postnatal microcephaly, micropthalmia, microcornia, bilateral congenital cataracts, short palpebral fissures, optic atrophy, severe mental retardation, and congenital hypotonia with subsequent spasticity. Only one patient had microcephaly at birth, highlighting the fact that congenital microcephaly is not a consistent feature of Micro syndrome. Analysis of the brain magnetic resonance imagings (MRIs) revealed a consistent pattern of polymicrogyria in the frontal and parietal lobes, wide sylvian fissures, a thin hypoplastic corpus callosum, and increased subdural spaces. All patients were homozygous for the mutations detected and all mutations were predicted to result in a truncated RAB3GAP1 protein. The analysis of nine polymorphic markers flanking the RAB3GAP1 gene showed that the mutation c.1410C>A (p.Tyr470X), for which a Danish patient was homozygous, occurred on a haplotype that is shared by the unrelated heterozygous parents of the patient. This suggests a possible founder effect for this mutation in the Danish population.
Similar content being viewed by others


Introduction
Warburg Micro Syndrome (WARBM, MIM 600118), also known as Micro Syndrome, is characterized by symptoms of microcephaly, mental retardation, corpus callosum hypoplasia, diffuse cortical or subcortical atrophy, congenital cataracts, microcornea, microphthalmia, progressive joint contractures with growth failure, and hypothalamic hypogonadism.1, 2 Micro Syndrome was first described by Warburg et al3 in a consanguineous Pakistani family with two affected sibs and an affected male cousin. Warburg et al3 delineated it from similar but distinct syndromes, such as Cerebrooculofacioskeletal Syndrome (COFS, MIM 214150), Cockayne Syndrome (CS; CSA, MIM 216400 and CSB MIM 133540), cataract, microcephaly, failure to thrive, Kyphoscoliosis syndrome (CAMAK and CAMFAK, MIM 212540), Martsolf Syndrome (MS, MIM 212720), lethal Rutledge syndrome (MIM 268670), and lethal Neu-Laxova syndrome (256520), all of which have microcephaly, severe mental retardation, childhood cataract, contractures, and genital hypoplasia as symptoms.
COFS syndrome shares many clinical and cellular similarities with CS in that cultured cells from patients with both these syndromes show hypersensitivity to ultraviolet radiation because of impaired nucleotide excision repair (NER).4, 5 Various forms of COFS and CS type A (CSA) and CS type B (CSB) have been found to be due to mutations in excision-repair cross-complementing genes (ERCC1, 2, 5, and 65, 6, 7, 8, 9). Impaired NER has not been shown in patients with Micro Syndrome.2 Micro Syndrome differs from COFS and CS in that patients have less prenatal and postnatal growth deficiency, cortical dysplasia rather than progressive brain atrophy, preservation of hearing, deficient visual perception despite early cataract removal, and a better prognosis for development and survival.2 Probably the most difficult differential diagnosis is between Micro syndrome and MS, which are primarily differentiated by the higher life expectancy of patients with MS syndrome.10
The molecular genetic basis of Micro Syndrome was unraveled by the finding of inactivating mutations in the Rab3 GTPase-activating protein, catalytic subunit gene (RAB3GAP1, chromosome 2q21.3, MIM *601536) in 12 of 14 families.11 Some of the Pakistani families described shared the same mutation.11 Micro Syndrome is extremely rare, and its true incidence is not known. To date, a total of nine different RAB3GAP1 mutations in patients from 14 different families have been described.11, 12, 13 Four of the mutations previously reported are recurrent and all the patients, with the exception of two from South Africa and Mexico, have consanguineous parents originating from the Middle East or Pakistan. In this study we provide a detailed clinical and molecular genetic analysis of seven patients from five families with Turkish, Palestinian, Guatemalan, and Danish ethnic backgrounds and five new mutations in RAB3GAP1. The parents in the Turkish and Palestinian families are reported to be consanguineous; however, the parents of the other two families are not closely related. The Danish patient is homozygous for a haplotype shared by the parents, suggesting a founder effect in the Danish population.
Materials and methods
Informed consent was obtained from the parents of all patients for the molecular genetic analysis of the patients, the publication of the clinical data, photos, and brain magnetic resonance imagings (MRIs).
DNA was extracted from EDTA blood using the Illustra BACC2 DNA extraction kit (GE Healthcare, Munich, Germany) and from buccal swabs from patient 6 using 5% Chelex 100 Resin (Bio-Rad, Munich, Germany).
PCR and DNA sequencing
The 24 coding exons of RAB3GAP1 and at least 30 bp of intronic sequence flanking the exons were amplified by PCR using Taq Polymerase (Qiagen, Hilden, Germany), and cycle sequenced using the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit Version 1.1 (Applied Biosystems, Darmstadt, Germany). Capillary electrophoresis was performed using an ABI 3100 sequencer (Applied Biosystems, Foster City, CA, USA). Sequence data were analyzed using SeqPilot DNA sequence analysis software (JSI, Kippenheim, Germany). The database sequence NM_012233.1 for RAB3GAP1 was used as reference sequence and PCR primers were developed in our laboratory (Supplementary Table 1).
cDNA analysis of the mutation c.648+5G>A in intron 7
RNA was extracted from lymphocytes that had been transformed with Ebstein–Barr virus, from patient 2 and his parents, using the RNeasy kit (Qiagen). Reverse transcription was performed using 1 μg RNA, an oligo DT Primer, and Superscript III (Invitrogen, Darmstadt, Germany). cDNA was PCR-amplified using primers RT-E5F and RT-E9R2 in exons 5 and 9, respectively, and the PCR product was sequenced using the same primers.
Haplotype analysis of polymorphic markers around RAB3GAP1
DNA from patient 5 and her parents was genotyped for nine microsatellite markers surrounding RAB3GAP1 on chromosome 2q21.3: D2S267, D2S368, AC110620GT24, AC016725TC23, AC020602CA17, AC012450TG23, AC012450AT37, D2S1334, and D2S2196. The primers used were according to Aligianis et al.11 The forward primer of each primer pair was labeled with 6-Fam and fragment analysis was performed through capillary electrophoresis on the ABI 3100 automatic sequencer, followed by analysis using the Genescan program (Applied Biosystems, USA).
Test for relatedness
The possible relatedness of the parents of patient 5, and those of the sibs 6 and 7, was tested using the PowerPlex16 Kit (Promega, Madison, WI, USA) for individual identity. The kit contains 15 polymorphic short tandem repeat (STR) markers located on 14 different chromosomes and an additional marker that shows different fragments for the X and Y chromosomes. Sample DNA (2 ng) was amplified in a multiplex PCR reaction in which the primers were labeled with one of three different fluorescent labels, and the resulting products were resolved on an ABI3100 automatic DNA sequencer. Fragment analysis and allele calling was performed using the Genescan Software (Applied Biosystems, USA).
Results
The clinical findings of the five patients are summarized in Supplementary Table 2. The individual patients are described below and the brain MRI findings are described collectively thereafter.
Patient 1
Patient 1 (Figure 1a) is the first child of consanguineous Turkish parents. She was born by cesarean section, due to the breech position, after a normal pregnancy. Ophthalmological evaluation showed bilateral microphthalmia and cataracts. Bilateral extirpation of the lenses was performed and glasses were prescribed. Further clinical evaluation showed axial hypotonia and peripheral hypertonia with rigidity of the lower limbs. Hypoplastic labia major were apparent in addition to some facial dysmorphic features, including low-set ears, a small mouth, retrognathia, and a prominent forehead with bitemporal hollowing. Microcephaly was not present at birth; however, it was apparent by age 2 years (between 10th and 25th centile). Laboratory investigations including TORCH serology and comparative genomic hybridization (array CGH) were all normal.

Photos of patients described in this study. (a) Patient 1 at age 2 years 4 months, (b) patient 6 at age 5 years 6 months, (c) patient 7 (sister of patient 6) at age 11 months, and (d–f) patient 2 at age 1 year (d) and at 4 years 9 months (e). The patient's overlapping toes are shown in f. (g) Profile of patient 3 at age 7 years. (h, i) Patient 5, with her hyperextended big toes shown in i. Consistent facial features appear to be low-set ears (apparent in a, d/e) a wide nasal bridge, which may be because of the patients’ small eyes (a, c, d/e), anteverted nares of variable severity (c, g), and a relatively narrow mouth with thin lips (a–c). Hypertrichosis is apparent in patients 1 and 6 (a, b).
Mutation analysis in RAB3GAP1 revealed the patient to be homozygous for the mutation c.1471C>T in exon 15 (Figure 2e), which results in the nonsense mutation, p.Arg491X. Both parents are heterozygous carriers of the mutation.

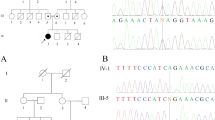
DNA sequence of the new RAB3GAP1 mutations found in this study. (a) Mutation c.264_270delAAGGATinsTTATTA in exon 4, found in patients 6 and 7; (b) mutation c.1410C>A in exon 15 in patient 5; (c, d) mutation c. 648+5G>A in intron 7, found in patient 2. The skipping of exon 7 in cDNA of patient 2 is shown in d. (e) Mutation c.1471C>T in exon 15 in patient 1 and (f) mutation c.1786_1789delAAAG in exon 17, found in patients 3 and 4.
Patient 2
This male patient (Figures 1d–f) is the first child of Turkish parents who are first cousins. The pregnancy was uncomplicated and routine ultrasound investigations did not reveal any abnormalities. There was suspicion of a CMV infection during the pregnancy; however, the child tested negative for CMV during the first week of life. The results of routine laboratory tests for newborn screening, including TORCH serology, EBV serology, and the Griffith test, were all normal. Routine chromosome analysis revealed a normal male karyotype.
Mutation analysis in RAB3GAP1 revealed homozygosity for the mutation c.648+5G>A in intron 7 (Figure 2c). Both parents of the patient are heterozygous carriers of the mutation. We performed RT-PCR on RNA from the patient and his parents, followed by PCR and cDNA sequencing using primers in exons 5 and 9. The transcript from the patient showed the complete skipping of exon 7 (Figure 2d), showing the lack of recognition of the donor splice site, as predicted by two splice prediction programs, ‘Splice view’ and ‘splice site prediction by neural network’, from the Berkeley Drosophila genome project (http://www.fruitfly.org/seq_tools/splice.html and www.fruitfly.org/seq_tools/splice.html). The transcripts from the parents were heterozygous for one normal transcript including exon 7 and one lacking exon 7. The loss of exon 7 eliminates amino acids 162 to 216 from the RAB3GAP1 transcript and is predicted to result in a frameshift and stop codon after five altered codons in exon 8.
Patients 3 and 4
The Palestinian parents of both these patients are first-degree cousins of Muslim origin. The paternal grandparents are also first cousins. Their oldest son is healthy and another son was born at 24 weeks of gestation and suffers from bronchopulmonary dysplasia and developmental delay, probably related to his premature birth.
In patient 3 (Figure 1g), bilateral cataracts were detected prenatally by ultrasound. At age 5 months he was operated on for cataract removal and glaucoma. At age 1 year, microphthalmia, microcephaly, hypotonia, and a small phallus were described. Eye examination at 2 years showed optic atrophy. At 7 years, he had severe mental retardation, quadriplegia, mild hearing loss, and reacted only to tactile stimulus. Chromosome analysis showed a normal male karyotype.
Patient 4 was found to have bilateral cataracts, a low nasal bridge, and flat forehead on ultrasound examination at 22 weeks of gestation. His head measurements were normal at this gestational age. He was suspected of having the same syndrome as his brother, and the pregnancy was terminated at 28 weeks of gestation. Mutation analysis in RAB3GAP1 in patients 3 and 4 revealed that they were both homozygous for the mutation c.1786_1789delAAAG in exon 17 (Figure 2f). The mutation results in a frameshift and stop codon: p.Lys596GlufsX613. The parents were not tested for the mutation.
Patient 5
Patient 5 (Figures 1h and i) is the first and only child of unrelated, Danish parents. The pregnancy and birth were unremarkable, and the female patient's eye abnormalities, that is, microphthalmia, microcornia, absent lens, and optic hypoplasia, were detected soon after birth. All routine postnatal screening, including TORCH serology and chromosome analysis, was normal. She failed to thrive, was hypotonic, and feeding through a gastric tube was necessary from age 3 months; the tube has subsequently been removed. She developed seizures at age 2 months, was treated with oxcarbazepin, and remained seizure free after the cessation of therapy. She remains severely hypotonic, with slight spasticity and severely impaired vision. She is unable to sit without support.
Mutation analysis in RAB3GAP1 revealed the patient to be homozygous for the mutation c.1410C>A in exon 15 (Figure 2b). The mutation is predicted to result in a stop codon, p.Tyr470X, and both parents were found to be heterozygous carriers of the mutation. As the parents reported that they were unrelated at least to the generation of their great grandparents, we decided to test whether the parents shared a common haplotype as the result of a possible founder effect in the Danish population. Haplotype analysis using nine microsatellite markers flanking RAB3GAP1 showed that the affected child was homozygous for a haplotype shared by the heterozygous parents (Figure 3). The analysis with 15 polymorphic STR markers (PowerPlex16 kit), located on chromosome arms other than chromosome 2q, showed that the parents shared only 6 of the 50 different alleles observed at the 15 loci (12%), thus confirming that the parents were not closely related.

Haplotype analysis of markers surrounding RAB3GAP1 in patient 5 and her parents. The allele carrying the c.1410C>A mutation is shown in italics.
Patients 6 and 7
Patients 6 (Figure 1b) and 7 (Figure 1c) are the first and second children of Spanish-speaking Guatemalan parents residing in the United States. The parents reported that they are unrelated; however, the same analysis as described above for the parents of patient 5, using 15 polymorphic STR markers, revealed that they shared 10 of the 42 observed alleles (23.8%), which may suggest that they are distantly related. Male patient 6 was clinically diagnosed with Micro Syndrome at 5 months. Pregnancy and delivery were unremarkable. Microcephaly, constricted pupils, excessive lacrimation, and microphallus were evident at 2.5 months. Postnatal testings, including TORCH serology, karyotyping, and analysis of the 607 BAC array, were normal. Cataract extraction was performed at 6 months. He showed failure to thrive with oral/pharyngeal dysphagia and required PEG feeding. He has cryptoorchidism and a micropenis, is severely mentally retarded, he does not visually track, and at age 3 years 7 months is unable to sit unsupported.
Micro Syndrome was suspected prenatally in patient 7 after the detection of bilateral cataracts. At age 1 year she shows profound developmental delay and cannot support her head. Mutation analysis in RAB3GAP1 in both patients 6 and 7 showed that both were homozygous for the insertion/deletion mutation: c.264_270delAAAGGATinsTTATTA in exon 4 (Figure 2a). The mutation results in a frameshift and stop codon, p.Lys89TyrfsX90. Both parents of the patients are heterozygous for the mutation.
Brain MRI findings
Selected images from brain magnetic resonance from all patients except patient 4 (fetus) are shown in Figure 4. The brains of all patients showed a relatively uniform combination of cortical malformations, including a wider than normal sylvian fissure, as shown in patients 1, 2, 3, and 5 (Figures 4a, c, e, i, and o), and polymicrogyria in the frontal and parietal regions (Figures 4c, g, k, o, and s). In patient 3, the extensive polymicrogyria extending over the frontal, parietal, and temporal lobes was most clearly apparent (Figure 4i), as was severe cerebellar hypoplasia (Figure 4j). All patients had a thin corpus callosum (Figures 4b, f, j, n, and r) with the rostrum and splenium absent in all patients except patient 3. Mild cerebellar vermis hypoplasia was apparent in patients 2 (Figure 4f), 5 (Figure 4n), and 6 (Figure 4r). All patients had increased subdural spaces, in particular the subarachnoid space on both temporal poles (Figures 4p and t) and delayed myelinization was apparent in patients 1, 2, and 5 (Figures 4c, g, and o).

Brain magnetic resonance images (MRIs) of patients 1–7, excluding patient 4 (fetus). Images a–d of patient 1 at age 6 months, images e–h of patient 2 at age 9 months, images i–l of patient 3 at age 7 years 3 months, images m–p of patient 5 at age 15 months, images q–t of patient 6 at age 3 months, images u–x of patient 7 (sister of patient 6) at postnatal day 1. A suitable parasaggital image was not available for patient 5. In images a, e, i, q, and u, the polymicrogyria around the frontal lobes, and in images a, e, i, and q, the wider-than-normal sylvian fissure is apparent. The sagittal sections b, f, j, n, r, and v show a consistently thin corpus callosum, shortened at the rostrum and genu (with the exception of patient 3, image j), cerebellar hypoplasia in patient 3 (j), and mild cerebellar vermis hypoplasia in patients 5– (images n, r, and v, respectively). The brainstem appears normal in all the patients. Increased subdural spaces are particularly apparent at the temporal poles, as shown in patients 5 and 7 (images m and x, respectively).
Discussion
One of the challenges for clinicians presented with patients with the combination of postnatal growth retardation, microcephaly, micropthalmia, cataracts, and hypogonadism (when apparent) is the differential diagnosis and the direction for molecular genetic analysis. To define what symptoms seem to be consistently present in patients with Micro Syndrome, we have extended the table of Graham et al2 with the clinical findings in previous cases of Micro Syndrome and our patients (Supplementary Table 3). Importantly, only one of our patients had microcephaly at birth. Microcephaly seemed to develop within the first year of life and was accompanied by general growth deficiency. Hence postnatal growth deficiency is a distinguishing feature of Micro Syndrome and this is consistent with the findings of Ainsworth et al14 and Graham et al.2 Two of the patients described by Mégarbané et al15 were microcephalic at birth; however, congenital microcephaly is not a consistent feature of Micro Syndrome. Micropthalmia, microcornia, bilateral congenital cataracts, short palpebral fissures, optic atrophy, severe mental retardation, and congenital hypotonia with subsequent spasticity were present in all patients. The above eye pathology, together with the soft features of deep set eyes, a wide nasal bridge, and relatively narrow mouth comprise common recognizable facial features. Hypogonadism was apparent in all male patients and one female patient; in the remaining two female patients, it was uncertain. Skeletal malformations were variable: hallux valgus and overextended and overlapping toes were reported in three of our patients and was shown in one of the patients reported by Graham et al.2 Clinodactyly, camptodactyly, and arachnodactyly have been reported in other patients.2, 12
Perhaps one of the most recognizable features of Micro Syndrome is the brain phenotype. All our patients showed a relatively consistent pattern of delayed myelinization, polymicrogyria of the frontal and parietal lobes, wide sylvian fissures, corpus callosum hypogenesis, and increased subdural spaces, particularly frontally and at the temporal poles. The brain has a particularly characteristic shape in axial slices, which is also apparent in the patients illustrated by Nassogne et al,16 Graham et al,2 and patients 2 and 6 shown by Abdel-Salam et al.12 Cerebellar vermis hypoplasia and cisterna magna were variably present.
Most mutations described to date have been found in Pakistani individuals, and two recurrent mutations have been identified, one of which (c.2801delC) appeared to occur on a common haplotype.11 Two additional mutations have recurred in Turkish patients (c.748+1G>A), and Lebanese and Egyptian patients (c.2011C>T). The previously reported mutations have all occurred in patients with consanguineous parents from Middle Eastern ethnic backgrounds, with the exception of a patient from South Africa whose ethnic background was not provided, and one patient from Mexico.11 The patients in this report are from diverse ethnic backgrounds and the parents of four of the patients are consanguineous. The parents of patients 6 and 7 are reported not to be consanguineous; however, they shared 10 of 42 (23.8%) of the different alleles observed between them for 15 different polymorphic STR markers on chromosome arms other than 2q. Although this is not an effective test for relatedness, the results indicate that the parents may be distantly related. However, the Danish parents of patient 5 shared only 6 of 50 (12%) of the different observed alleles between them and are certain that their great grandparents were not related. They nevertheless shared a common haplotype for markers surrounding the RAB3GAP1 gene, for which their affected daughter is homozygous, thus suggesting a founder effect for the mutation c.1410C>A in the Danish population. Such a founder effect has previously been reported in the Danish population for a mutation in the OPA1 gene in patients with dominant optic atrophy,17 for MLH1 and MSH6 mutations in Lynch Syndrome,18 and the Cys282Tyr HFE mutation in hereditary hemochromatosis.19 A higher incidence of Micro Syndrome in the Danish population has so far not been reported; however, the targeted analysis of the c.1410C>A mutation in further Danish patients with suspected Micro Syndrome should help to validate our finding.
The Rab family of small G proteins are regarded as principal classes of GTPases that have an important role in the regulation of intracellular vesicle transport.20, 21 The Rab3 subfamily is associated with secretory granules or vesicles, and has a crucial role in regulated exocytosis.20, 22, 23 Rab3A is the most abundant isoform in brain, in which it is localized on synaptic vesicles, and evidence is accumulating that it has a key role in neurotransmitter release and synaptic plasticity.24 RAB3GAP, which converts active Rab3-GTP to inactive Rab3-GDP, thereby regulating Rab3 activity, is a heterodimeric complex consisting of a 130 kDa catalytic subunit encoded by RAB3GAP1, and a 150 kDa non-catalytic subunit RAB3GAP2.25, 26 The catalytic activity of RAB3GAP1 is localized in the C-terminal region (codons 601–98125). The mutations in RAB3GAP1 that have so far been detected in patients with Micro Syndrome are all predicted to result in stop codons and are distributed throughout the coding region from codons 89 to 934 (Aligianis et al11 and this study). Therefore, all the mutations are predicted to result in a truncated protein either before or within the region important for catalytic activity. This is in line with the prediction that most RAB3GAP1 mutations produce a mutant protein without catalytic activity.11 This may explain why there seems to be relatively little phenotypic variation between the patients so far described with different mutations in RAB3GAP1. A single splice mutation has been found in RAB3GAP2 in three sibs of a consanguineous Pakistani family with Martsolf Syndrome.10 These patients are able to walk and talk and show much milder ocular and neurodevelopmental defects than those with Micro Syndrome and mutations in RAB3GAP1.10 Currently, it is still not clear whether or not the milder phenotype is because of the nature of the mutation.
There may be additional genes that, when mutated, cause Micro Syndrome, as linkage to RAB3GAP1 and RAB3GAP2 has been excluded in families of patients with suspected Micro Syndrome, and kindreds with or without mutations in RAB3GAP1 were clinically indistinguishable.11, 12 However, the addition of the seven patients with Micro Syndrome and mutations in RAB3GAP1 in this report to the previously described patients with a diagnosis of Micro Syndrome provides an even clearer picture of the symptoms essential to Micro syndrome.
References
Derbent M, Agras PI, Gedik S, Oto S, Alehan F, Saatci U : Congenital cataract, microphthalmia, hypoplasia of corpus callosum and hypogenitalism: report and review of Micro syndrome. Am J Med Genet A 2004; 128A: 232–234.
Graham Jr JM, Hennekam R, Dobyns WB, Roeder E, Busch D : MICRO syndrome: an entity distinct from COFS syndrome. Am J Med Genet A 2004; 128A: 235–245.
Warburg M, Sjo O, Fledelius HC, Pedersen SA : Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am J Dis Child 1993; 147: 1309–1312.
Meira LB, Graham Jr JM, Greenberg CR et al: Manitoba aboriginal kindred with original cerebro-oculo-facio-skeletal syndrome has a mutation in the Cockayne syndrome group B (CSB) gene. Am J Hum Genet 2000; 66: 1221–1228.
Graham Jr JM, Anyane-Yeboa K, Raams A et al: Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet 2001; 69: 291–300.
Jaspers NG, Raams A, Silengo MC et al: First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet 2007; 80: 457–466.
Cleaver JE, Thompson LH, Richardson AS, States JC : A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat 1999; 14: 9–22.
Nouspikel T, Lalle P, Leadon SA, Cooper PK, Clarkson SG : A common mutational pattern in Cockayne syndrome patients from xeroderma pigmentosum group G: implications for a second XPG function. Proc Natl Acad Sci USA 1997; 94: 3116–3121.
Mallery DL, Tanganelli B, Colella S et al: Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am J Hum Genet 1998; 62: 77–85.
Aligianis IA, Morgan NV, Mione M et al: Mutation in Rab3 GTPase-activating protein (RAB3GAP) noncatalytic subunit in a kindred with Martsolf syndrome. Am J Hum Genet 2006; 78: 702–707.
Aligianis IA, Johnson CA, Gissen P et al: Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat Genet 2005; 37: 221–223.
Abdel-Salam GM, Hassan NA, Kayed HF, Aligianis IA : Phenotypic variability in Micro syndrome: report of new cases. Genet Couns 2007; 18: 423–435.
Yuksel A, Yesil G, Aras C, Seven M : Warburg Micro syndrome in a Turkish boy. Clin Dysmorphol 2007; 16: 89–93.
Ainsworth JR, Morton JE, Good P et al: Micro syndrome in Muslim Pakistan children. Ophthalmology 2001; 108: 491–497.
Mégarbané A, Choueiri R, Bleik J, Mezzina M, Caillaud C : Microcephaly, microphthalmia, congenital cataract, optic atrophy, short stature, hypotonia, severe psychomotor retardation, and cerebral malformations: a second family with micro syndrome or a new syndrome? J Med Genet 1999; 36: 637–640.
Nassogne MC, Henrot B, Saint-Martin C, Kadhim H, Dobyns WB, Sebire G : Polymicrogyria and motor neuropathy in Micro syndrome. Neuropediatrics 2000; 31: 218–221.
Thiselton DL, Alexander C, Morris A et al: A frameshift mutation in exon 28 of the OPA1 gene explains the high prevalence of dominant optic atrophy in the Danish population: evidence for a founder effect. Hum Genet 2001; 109: 498–502.
Nilbert M, Wikman FP, Hansen TV et al: Major contribution from recurrent alterations and MSH6 mutations in the Danish Lynch syndrome population. Fam Cancer 2009; 8: 75–83.
Milman N, Pedersen P : Evidence that the Cys282Tyr mutation of the HFE gene originated from a population in Southern Scandinavia and spread with the Vikings. Clin Genet 2003; 64: 36–47.
Takai Y, Sasaki T, Matozaki T : Small GTP-binding proteins. Physiol Rev 2001; 81: 153–208.
Zerial M, McBride H : Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2001; 2: 107–117.
Geppert M, Sudhof TC : RAB3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci 1998; 21: 75–95.
Takai Y, Sasaki T, Shirataki H, Nakanishi H : Rab3A small GTP-binding protein in Ca(2+)-dependent exocytosis. Genes Cells 1996; 1: 615–632.
Sakane A, Miyoshi J, Takai Y, Sasaki T : Analysis on the emerging role of Rab3 GTPase-activating protein in Warburg Micro and Martsolf syndrome. Methods Enzymol 2008; 438: 131–139.
Fukui K, Sasaki T, Imazumi K, Matsuura Y, Nakanishi H, Takai Y : Isolation and characterization of a GTPase activating protein specific for the Rab3 subfamily of small G proteins. J Biol Chem 1997; 272: 4655–4658.
Nagano F, Sasaki T, Fukui K, Asakura T, Imazumi K, Takai Y : Molecular cloning and characterization of the noncatalytic subunit of the Rab3 subfamily-specific GTPase-activating protein. J Biol Chem 1998; 273: 24781–24785.
Acknowledgements
We are grateful to the parents of the patients in this study for their willing cooperation and to Angela Steiert for technical assistance. We thank Dr Beate Albrecht, Universitätsklinikum Essen, Germany, for motivating our work on Micro Syndrome.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Morris-Rosendahl, D., Segel, R., Born, A. et al. New RAB3GAP1 mutations in patients with Warburg Micro Syndrome from different ethnic backgrounds and a possible founder effect in the Danish. Eur J Hum Genet 18, 1100–1106 (2010). https://doi.org/10.1038/ejhg.2010.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.79
Keywords
This article is cited by
-
Knockout mouse models as a resource for the study of rare diseases
Mammalian Genome (2023)
-
Novel manifestations of Warburg micro syndrome type 1 caused by a new splicing variant of RAB3GAP1: a case report
BMC Neurology (2021)
-
Two novel Warburg micro syndrome 1 cases caused by pathogenic variants in RAB3GAP1
Human Genome Variation (2021)
-
Genetics in Keratoconus: where are we?
Eye and Vision (2016)
-
Two novel homozygous RAB3GAP1 mutations cause Warburg micro syndrome
Human Genome Variation (2015)
