
Abstract
The potentially functional polymorphism, rs763110 (−844C>T), in the promoter region of the FAS ligand (FASL) gene, has been implicated in cancer risk, but individually published studies show inconclusive results. To derive a more precise estimation of the association between the FASL rs763110 and risk of cancer, we performed a meta-analysis of 19 published studies that included 11 105 cancer cases and 11 372 controls. We used odds ratios (ORs) and 95% confidence intervals (CIs) to assess the strength of the associations. Overall, the rs763110 CT and TT variant genotypes were associated with a significantly reduced cancer risk of all cancer types in different genetic models (homozygote comparison: OR=0.80, 95% CI: 0.68–0.95, Pheterogeneity=0.001; heterozygote comparison: OR=0.82, 95% CI: 0.72–0.95, Pheterogeneity<0.001; dominant model comparison: OR=0.82, 95% CI: 0.71–0.94, Pheterogeneity<0.001; and recessive model comparison: OR=0.88, 95% CI: 0.81–0.96, Pheterogeneity=0.074). In the stratified analyses, the risk remained for studies of the smoking-related cancers and Asian populations, or population-based studies in all the genetic models. Although some modest bias could not be eliminated, this meta-analysis suggests that the FASL rs763110 T allele has a possible protective effect on cancer risk.
Similar content being viewed by others


Introduction
One of the major functions of human immune system is to eliminate potentially malignant cells, for which anticancer T lymphocytes play a crucial role in immunosurveillance of cancer cells. However, T lymphocytes can be triggered into an apoptosis process, also called activation-induced cell death (AICD).1 Many studies indicate that AICD is an important mechanism responsible for the increased apoptosis rate among the tumor-infiltrating lymphocytes, leading to the transformed cells escaping the elimination by anticancer immunosurveillance and, therefore, AICD contributes to cancer development.2, 3
Activation-induced cell death is a FAS ligand (FASL)-dependent process, and the increased expression of FASL can cause AICD in peripheral T cells.4 Antigenic stimulation of cancer within the tumor microenvironment can induce tumor-infiltrating lymphocytes to overexpress FASL and subsequently lead to suicide or fratricide of tumor-infiltrating lymphocytes through the FAS–FASL interaction-triggered apoptosis.5 In addition, mutations in death-receptor pathway genes may cause a reduced expression of FAS, but an increased expression of FASL, in cancers.6, 7 Decreased expression of FAS may protect transformed cells from the elimination by anticancer immuneresponses, whereas increased FASL expression may enhance the ability of cancer cells to counterattack the immune system by killing FAS-sensitive lymphocytes, thereby contributing to cancer development.8, 9
The human FASL gene (GenBank accession no: Z96050) is located on chromosome 1q23 and comprised of four exons. FASL can trigger cell-death signal cascade by crosslinking with FAS, and the FAS/FASL system plays a crucial role in modulating apoptosis and maintaining the homeostasis.10 As shown in the HapMap and dbSNP databases, the FASL gene is highly polymorphic, but the most extensively studied polymorphism is the C to T substitution at position −844 (−844C>T, rs763110) in the promoter region of the FASL gene. This potentially functional polymorphism is located within a putative binding motif of CAAT/enhancer-binding protein β (C/EBP-β) transcription factor, and a considerably higher basal expression of FASL is associated with the FASL rs763110 C allele compared with the T allele.11 As for other SNPs in the FASL gene, such as rs5030772,12 there are relatively fewer studies reported to date. Thus, the sample size for these SNPs is too small for a meaningful meta-analysis. Therefore, we chose to perform a meta-analysis of the rs763110.
Recently, many studies have investigated the role of the FASL rs763110 in the etiology of cancers of various organs, including the breast,13, 14, 15 cervix,16, 17, 18, 19 bladder,20 lung,21, 22, 23 head and neck,12 skin,24, 25 esophagus,26 stomach,27 ovary,28 thymus,29 and pancreas.30 However, the results of these studies remain inconclusive. In this report, we performed a meta-analysis to estimate the effect of this polymorphism on cancer risk as well as to quantify the potential between-study heterogeneity.
Materials and methods
Identification and eligibility of relevant studies
Two online electronic databases (PubMed and Embase) were searched (the last search update was 1 February 2009, using the search terms ‘FAS ligand’, ‘FASL’, ‘TNFSF6’, ‘CD95L’, ‘polymorphism’, and ‘cancer’). The search was limited to English-language papers. We also used the PubMed option ‘Related Articles’ in each research article to search potentially relevant articles. Moreover, references of all included articles were also screened. Of the studies with the same or overlapping data published by the same investigators, we selected the most recent ones with the largest number of participants. Studies included in our meta-analysis have to meet the following inclusion criteria: (1) use a case–control design and (2) contain available genotype frequency. Major reasons for exclusion of studies were: (1) no control population; (2) duplicate of earlier publication; and (3) no usable genotype frequency data.
Data extraction
Information was carefully extracted from all eligible publications independently by two of the authors, according to the inclusion criteria. Discrepancies were adjudicated by a third reviewer until consensus was achieved on every item. For each study, the following characteristics were collected: the first author's last name, year of publication, country of origin, ethnicity, matching conditions, numbers of genotyped cases and controls, source of control groups (population- or hospital-based controls), source of DNA, genotyping methods, and quality control. Different ethnic descents were categorized as European, Asian, and African.
Statistical analysis
The strength of the association between the FASL rs763110 and cancers was measured by odds ratios (ORs) with 95% confidence intervals (CIs). The statistical significance of the pooled OR was determined using the Z-test. We first estimated cancer risks associated with the CT and TT genotypes, compared with the wild-type CC homozygote, and then evaluated the risks associated with (TT/CT) vs CC and TT vs (CT/CC), assuming the dominant and recessive effects of the variant T allele, respectively. Stratification analyses were also performed by cancer types (if one cancer type contained less than three individual studies, it was combined into the ‘other cancers’ group), ethnicity, and source of controls. Lung, bladder, esophageal, head and neck, and pancreatic cancers were defined as smoking-related cancers, because tobacco smoking is an established risk factor for these cancers.26, 31, 32, 33, 34 In addition, given the roles of estrogen level in the etiology of breast, cervical, and ovarian cancers, these cancers were defined as estrogen-related cancers.35, 36 Then we examined whether the FASL rs763110 was associated with the risk of these cancers.
Heterogeneity assumption was checked by the χ2-based Q-test and considered significant when P<0.05. Both fixed-effects model using the Mantel–Haenszel method37 and random-effects model using the DerSimonian and Laird method38 were used to pool the results. Furthermore, the variables of ethnicities, cancer sites, sources of controls, and sample sizes were examined in a meta-regression model to explore the possible heterogeneity among different kinds of studies. The interstudy variance (τ2) was used to quantify the degree of heterogeneity between studies, and the percentage of τ2 was used to describe the extent of explained heterogeneity of the characteristics.39 Sensitivity analyses were also performed to assess the stability of the results. Funnel plots and Egger's linear regression test were used to provide diagnosis of potential publication bias.40 All analyses were carried out with SAS (version 9.1; SAS Institute Inc., Cary, NC, USA) and Stata software (version 8.2; StataCorp. LP, College Station, TX, USA), using two-sided P-values.
Results
Characteristics of studies
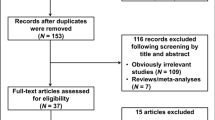
There were 35 eligible studies as a result of the search and screening carried out on the basis of our eligibility criteria. During the extraction of data, 16 articles were excluded, because they did not provide allele frequencies needed for OR calculation, leaving 19 eligible studies that had assessed the association between rs763110 and cancer risk using human genomic DNA samples. Among the 19 eligible studies, there were 11 105 cases with different cancer types and 11 372 controls. The characteristics of selected studies are summarized in Table 1. All studies were case–control studies, including four cervical cancer studies, three breast cancer studies, three lung cancer studies, and the others, which were categorized into the ‘other cancers’ group. There were 10 studies of Asian descendents and nine studies of European descendents. Cancers were confirmed histologically or pathologically in most studies. Of the 19 studies, 16 studies used frequency-matched controls to the cases by the age, sex, or ethnicity. A classic PCR–restriction fragment length polymorphism (PCR-RFLP) assay was performed in 14 of the 19 studies; however, only 13 studies mentioned quality control of the genotyping, such as blindness to the case–control status, randomly repeated assays, or validation using a different genotyping method. Besides, 15 studies investigated the interactions between the FASL rs763110 and environmental factors or other genes such as FAS. The distribution of genotypes in the controls of all studies was in agreement with Hardy–Weinberg equilibrium except for one study by Sun et al.16
Quantitative synthesis
We observed a wide variation of the rs763110 T-allele frequencies across different ethnicities. The average T-allele frequencies in Asian and European population were 0.29 and 0.36, respectively, which is very close to that obtained from the HapMap Project (0.30 for CHB and 0.36 for CEU). The best genetic model was determined according to Thakkinstian's criteria,41 and the results suggested that the genetic model was most likely to be dominant. Overall, there was evidence of an association between the reduced cancer risk and the variant genotypes in different genetic models when all the eligible studies were pooled into the meta-analysis. Compared with the wild-type homozygote genotype, CC, the variant genotypes, CT and TT, were associated with a statistically significant decreased risk of all types of cancer (OR=0.82, 95% CI: 0.72–0.95, Pheterogeneity<0.001 for CT; OR=0.80, 95% CI: 0.68–0.95, Pheterogeneity=0.001 for TT; Table 2). In addition, significant main effects were also observed both in dominant and recessive models (dominant model: OR=0.82, 95% CI: 0.71–0.94, Pheterogeneity<0.001; recessive model: OR=0.88, 95% CI: 0.81–0.96, Pheterogeneity=0.074; Table 2, Figure 1). Notably, the T-variant genotypes (CT and TT) were associated with a decreased cancer risk in a dose-response manner compared with the CC genotype (OR=0.82, 95% CI: 0.72–0.95 for CT and 0.80, 0.68–0.95 for TT; Ptrend<0.001).

Forest plot of cancer risk associated with the FASL rs763110 polymorphism (TT/CT vs CC). The squares and horizontal lines correspond to the study-specific OR and 95% CI. The area of the squares reflects the study-specific weight (inverse of the variance). The diamond represents the pooled OR and 95% CI.
In the stratified analysis by cancer type, as shown in Table 2, we found that individuals with the variant genotypes had a significantly lower risk of the ‘other cancers’ in different genetic models, but not of cervical, breast, or lung cancers. Besides, significantly reduced risk was also found for the variant genotypes in different genetic models among the smoking-related cancers (Figure 2). For estrogen-related cancers, significantly reduced risk was observed only in the homozygote comparison (OR=0.82, 95% CI: 0.70–0.96, Pheterogeneity=0.078; Table 2).

Forest plot of smoking-related cancers risk associated with the FASL rs763110 polymorphism (TT/CT vs CC). The squares and horizontal lines correspond to the study-specific OR and 95% CI. The area of the squares reflects the study-specific weight (inverse of the variance). The diamond represents the pooled OR and 95% CI.
In the subgroup analysis by ethnicity, significantly decreased risks were also found among Asian populations in all models tested (homozygote comparison: OR=0.62, 95% CI: 0.53–0.73, Pheterogeneity=0.103; heterozygote comparison: OR=0.70, 95% CI: 0.59–0.84, Pheterogeneity<0.001; dominant model: OR=0.69; 95% CI: 0.59–0.82, Pheterogeneity<0.001; recessive model: OR=0.73; 95% CI: 0.63–0.86, Pheterogeneity=0.287). However, these similar significant associations were not observed for European populations (Table 2, Figure 1).
Then, we divided these studies into two subgroups according to their sources of controls. For the studies with population-based controls, we found that the variant genotypes were associated with a significantly decreased cancer risk in all genetic models (homozygote comparison: OR=0.72, 95% CI: 0.54–0.95, Pheterogeneity<0.001; heterozygote comparison: OR=0.77, 95% CI: 0.63–0.95, Pheterogeneity<0.001; dominant model: OR=0.77, 95% CI: 0.62–0.94, Pheterogeneity<0.001; recessive model: OR=0.80, 95% CI: 0.65–0.99, Pheterogeneity=0.014). However, among studies with hospital-based controls, no significant association was observed in any genetic model (Table 2, Figure 3). By comparing with the variant T-allele frequency in controls between population- and hospital-based studies, we found that the T-allele frequency among controls in hospital-based studies (33.5%) was slightly higher than those in population-based studies (31.6%). These data suggested that those studies whose controls were selected from hospital may underestimate the cancer risk.

Forest plot of cancer risk among studies with population-based controls associated with the FASL rs763110 polymorphism (TT/CT vs CC). The squares and horizontal lines correspond to the study-specific OR and 95% CI. The area of the squares reflects the study-specific weight (inverse of the variance). The diamond represents the pooled OR and 95% CI.
Test of heterogeneity
There was significant heterogeneity for homozygote comparison (TT vs CC: Pheterogeneity=0.001), heterozygote comparison (CT vs CC: Pheterogeneity<0.001), and dominant model comparison (TT/CT vs CC: Pheterogeneity<0.001), but not for recessive model comparison (TT vs CT/CC: Pheterogeneity=0.074). Then, we assessed the source of heterogeneity for dominant model comparison by cancer site, ethnicity, source of controls, sample size (participants more than 500 in both cases and controls), and genotyping method. As a result, ethnicity (P<0.001), cancer site (P=0.024), and genotyping method (P<0.001), but not the source of controls and sample size, were found to contribute to the substantial heterogeneity. Moreover, meta-regression analyses indicated that, in dominant model comparison, ethnicity could explain 69.3% of the τ2, whereas cancer sites could explain 4.71% of the τ2.
Sensitivity analyses
Sensitivity analyses indicated that two independent studies by Park et al22 and Kang et al18 were the main origin of heterogeneity in Asians. The heterogeneity was effectively decreased or removed after exclusion of these two studies (TT vs CC: Pheterogeneity=0.320 and TT/CT vs CC: Pheterogeneity=0.05). Although the genotype distribution in the study by Sun et al16 did not follow Hardy–Weinberg equilibrium, the corresponding pooled ORs were not materially altered with (dominant model: OR=0.82, 95% CI: 0.71–0.94) or without (dominant model: OR=0.84, 95% CI: 0.73–0.97) including this study. Similarly, no other single study influenced the pooled OR qualitatively, as indicated by sensitivity analyses, suggesting that the results of this meta-analysis are stable.
Publication bias
Begg's funnel plot and Egger's test were performed to assess the publication bias of literatures. The shapes of the funnel plots did not reveal any evidence of obvious asymmetry in all comparison models. Then, the Egger's test was used to provide statistical evidence of funnel plot symmetry. The results still did not show any evidence of publication bias (t=−0.70, P=0.496 for TT/CT vs CC; Figure 4).

Begg’s funnel plot for publication bias test (TT/CT vs CC). Each point represents a separate study for the indicated association. log[OR], natural logarithm of odds ratio. Horizontal line, mean effect size.
Discussion
This meta-analysis, including 11 105 cases and 11 372 controls from 19 published case–control studies, explored the association between a potentially functional polymorphism, rs763110, within the FASL promoter region and cancer risk. We found that there was evidence that the variant genotypes of the rs763110 were associated with a significant decrease in overall cancer risk in a T-allele dose-response manner. This finding is biologically plausible.
Immune surveillance of T cells plays an essential role in maintaining homeostasis,1, 42 and the AICD process of T cells is regulated by the FAS/FASL system. Genetic variations of crucial genes in this cell-death pathway may thus influence susceptibility to cancers. The FASL rs763110 has been shown to have a substantial impact on the promoter activity of the FASL gene in an in vitro assay system, because the variant affects a binding motif for the transcription factor C/EBP. A considerably higher basal expression of FASL is associated with the FASL rs763110 C allele compared with the T allele.11 It has been shown that the FASL rs763110 C allele, which had a higher expression of FASL in T cells, was associated with an enhanced rate of AICD in T cells, suggesting that the rs763110 C allele may be a risk allele for cervical cancer.16 AICD of T lymphocytes may help malignant cells to escape from killing by natural killing cells.1, 3 The C to T change at the FASL rs763110 polymorphic site strongly reduces the expression of FASL, and consequentially reduces AICD of tumor-specific T cells in response to malignant cells, which may result in more powerful immune surveillance and reduce susceptibility to cancer risk among individuals carrying the T allele.16 In our present meta-analysis, we found that individuals with the TT genotype were associated with a lower cancer risk than participants with the CC genotype, which was consistent with experimental findings.
Tobacco smoke contains hundreds of chemicals some of which are carcinogens, such as polycyclic aromatic hydrocarbons and N-nitroso compounds, as evidenced in animal studies.43 Tobacco smoking is a well-established risk factor for cancers of many organs, including lung, bladder, esophagus, head and neck, and pancreas.26, 31, 32, 33, 34 Besides, studies have also shown that chronic smoking could enhance the FASL expression in peripheral blood lymphocytes, which is believed to impair the immune function in smokers.44 Moreover, transformed cells resulting from exposure to tobacco carcinogens may evade tumor-infiltrating lymphocyte killing to become malignant because of high expression of FASL.45
Although we found an evidence for the association between the FASL rs763110 T-variant genotypes and the reduced risk of smoking-related cancers, this was not evident for other cancers. This difference may be due to limited statistical power as a result of the small sample size and low-penetrance effects of this SNP in these non-smoking-related cancers as well as possibly different carcinogenic mechanisms underlying the etiology. Therefore, it is possible that the FASL rs763110 may exert varying effects on different cancers. For example, the development of both cervical cancer and breast cancer may involve hormonal factors such as estrogen level and reproductive events (that is, early menarche, late menopause, and nulliparity).46 Estrogen may increase the mitotic activity of the cells and the number of DNA replication errors.36, 47, 48 Mor et al49 showed that FASL in breast tissue was functionally active and that FASL expression was regulated by estrogen, possibly as a result of a putative estrogen receptor response element in the FASL promoter region. Estrogen has also been shown to trigger T-cell apoptosis by upregulating FAS and FASL in cancer cells.50 Therefore, the effect of estrogen level on the risk may outweigh that of this FASL rs763110 in estrogen-related cancers. However, more studies are needed to validate the results of this meta-analysis for estrogen-related cancers.
We found an evidence for the association between the FASL rs763110 and cancer risk among Asians, but not Europeans, a possible reflection of differences in genetic background and gene–environment interactions in the etiology. For example, the T-allele frequency among controls was 0.29 in Asian populations and 0.36 in European populations, suggesting a possible ethnic difference. In addition, half (5/10) of the Asian studies focused on smoking-related cancers, whereas only two studies12, 23 investigated smoking-related cancer in the European populations. However, there was no reported study in the African populations. Therefore, additional studies are warranted to further validate possible ethnic differences in the effect of this functional SNP on cancer risk.
We also found that the association was significant among studies using the population-based controls, but not the hospital-based controls. This may be because the hospital-based studies have inherent selection biases due to the fact that such controls may not be representative of the study population or the general population, particularly when the genotypes under investigation were associated with the disease-related conditions that hospital-based controls may have. Thus, the use of proper and representative population-based control participants is very important in reducing biases in such genotype association studies.
In this meta-analysis, three studies assessed the SNPs in the adjacent regions that were in linkage disequilibrium with rs76311012, 15, 25 in which FASL haplotype and combined genotype analyses showed that the variant haplotypes were associated with significantly higher cancer risk. These results indicate that FASL polymorphisms may play a crucial role in the development of cancer, which is biologically plausible. In future, large, well-designed studies for haplotypes, along with combined analysis, are recommended.
Identifying the source of heterogeneity is one of the most important goals of the meta-analysis. Thus, we stratified the studies according to ethnicity, cancer type, source of controls, sample size, and genotyping method, and we found that several factors, such as ethnicity, cancer type, and genotyping method, may have contributed to the observed heterogeneity, which was further confirmed by meta-regression analyses.
Some limitations of this meta-analysis should be addressed. First, lack of the original data of the reviewed studies limited our further evaluation of potential interactions, because the interactions between gene–gene, gene–environment, and even different polymorphic loci of the same gene may modulate cancer risk. Second, misclassifications on disease status and genotypes may also influence the results, because cases in several studies were not confirmed by pathology or other gold standard methods, and the quality control of genotyping was also not well-documented in some studies. Third, the numbers of published studies were not sufficiently large for a comprehensive analysis, particularly for any given cancer site. In spite of these, our meta-analysis also had some advantages. First, substantial number of cases and controls were pooled from different studies, which significantly increased statistical power of the analysis. Second, the quality of case–control studies included in the current meta-analysis was satisfactory based on our selection criteria. Third, we did not detect any publication bias indicating that the whole pooled result may be unbiased.
In summary, this meta-analysis identified an evidence of the association between the FASL rs763110 and cancer risk, supporting the hypothesis that the FASL rs763110 may be a low-penetrance susceptibility marker of cancer. Our results also suggest that additional large studies are warranted to validate possible ethnic difference in the risk. Future studies should use standardized unbiased genotyping methods, homogeneous cancer patients, and well-matched controls with multi-ethnic groups. Moreover, gene–gene and gene–environment interactions should also be examined in the future analysis. These future studies should lead to better, comprehensive understanding of the association between the FASL rs763110 and cancer risk.
Accession codes
References
Green DR, Droin N, Pinkoski M : Activation-induced cell death in T cells. Immunol Rev 2003; 193: 70–81.
Maher S, Toomey D, Condron C, Bouchier-Hayes D : Activation-induced cell death: the controversial role of Fas and Fas ligand in immune privilege and tumour counterattack. Immunol Cell Biol 2002; 80: 131–137.
Chappell DB, Restifo NP : T cell-tumor cell: a fatal interaction? Cancer Immunol Immunother 1998; 47: 65–71.
Ju ST, Panka DJ, Cui H et al: Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 1995; 373: 444–448.
Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA : Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995; 270: 1189–1192.
O’Connell J, O’Sullivan GC, Collins JK, Shanahan F : The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med 1996; 184: 1075–1082.
Crnogorac-Jurcevic T, Efthimiou E, Capelli P et al: Gene expression profiles of pancreatic cancer and stromal desmoplasia. Oncogene 2001; 20: 7437–7446.
Niehans GA, Brunner T, Frizelle SP et al: Human lung carcinomas express Fas ligand. Cancer Res 1997; 57: 1007–1012.
Bennett MW, O’Connell J, O’Sullivan GC et al: The Fas counterattack in vivo: apoptotic depletion of tumor-infiltrating lymphocytes associated with Fas ligand expression by human esophageal carcinoma. J Immunol 1998; 160: 5669–5675.
Houston A, O’Connell J : The Fas signalling pathway and its role in the pathogenesis of cancer. Curr Opin Pharmacol 2004; 4: 321–326.
Wu J, Metz C, Xu X et al: A novel polymorphic CAAT/enhancer-binding protein beta element in the FasL gene promoter alters Fas ligand expression: a candidate background gene in African American systemic lupus erythematosus patients. J Immunol 2003; 170: 132–138.
Zhang Z, Wang LE, Sturgis EM et al: Polymorphisms of FAS and FAS ligand genes involved in the death pathway and risk and progression of squamous cell carcinoma of the head and neck. Clin Cancer Res 2006; 12: 5596–5602.
Crew KD, Gammon MD, Terry MB et al: Genetic polymorphisms in the apoptosis-associated genes FAS and FASL and breast cancer risk. Carcinogenesis 2007; 28: 2548–2551.
Krippl P, Langsenlehner U, Renner W, Koppel H, Samonigg H : Re: Polymorphisms of death pathway genes FAS and FASL in esophageal squamous-cell carcinoma. J Natl Cancer Inst 2004; 96: 1478–1479; author reply 1479.
Zhang B, Sun T, Xue L et al: Functional polymorphisms in FAS and FASL contribute to increased apoptosis of tumor infiltration lymphocytes and risk of breast cancer. Carcinogenesis 2007; 28: 1067–1073.
Sun T, Zhou Y, Li H et al: FASL −844C polymorphism is associated with increased activation-induced T cell death and risk of cervical cancer. J Exp Med 2005; 202: 967–974.
Lai HC, Lin WY, Lin YW et al: Genetic polymorphisms of FAS and FASL (CD95/CD95L) genes in cervical carcinogenesis: an analysis of haplotype and gene-gene interaction. Gynecol Oncol 2005; 99: 113–118.
Kang S, Dong SM, Seo SS, Kim JW, Park SY : FAS −1377 G/A polymorphism and the risk of lymph node metastasis in cervical cancer. Cancer Genet Cytogenet 2008; 180: 1–5.
Ivansson EL, Gustavsson IM, Magnusson JJ et al: Variants of chemokine receptor 2 and interleukin 4 receptor, but not interleukin 10 or Fas ligand, increase risk of cervical cancer. Int J Cancer 2007; 121: 2451–2457.
Li C, Wu W, Liu J et al: Functional polymorphisms in the promoter regions of the FAS and FAS ligand genes and risk of bladder cancer in south China: a case-control analysis. Pharmacogenet Genomics 2006; 16: 245–251.
Zhang X, Miao X, Sun T et al: Functional polymorphisms in cell death pathway genes FAS and FASL contribute to risk of lung cancer. J Med Genet 2005; 42: 479–484.
Park SH, Choi JE, Kim EJ et al: Polymorphisms in the FAS and FASL genes and risk of lung cancer in a Korean population. Lung Cancer 2006; 54: 303–308.
Ter-Minassian M, Zhai R, Asomaning K et al: Apoptosis gene polymorphisms, age, smoking and the risk of non-small cell lung cancer. Carcinogenesis 2008; 29: 2147–2152.
Zhang H, Sun XF, Synnerstad I, Rosdahl I : Importance of FAS-1377, FAS-670, and FASL-844 polymorphisms in tumor onset, progression, and pigment phenotypes of Swedish patients with melanoma: a case-control analysis. Cancer J 2007; 13: 233–237.
Li C, Larson D, Zhang Z et al: Polymorphisms of the FAS and FAS ligand genes associated with risk of cutaneous malignant melanoma. Pharmacogenet Genomics 2006; 16: 253–263.
Sun T, Miao X, Zhang X, Tan W, Xiong P, Lin D : Polymorphisms of death pathway genes FAS and FASL in esophageal squamous-cell carcinoma. J Natl Cancer Inst 2004; 96: 1030–1036.
Hsu PI, Lu PJ, Wang EM et al: Polymorphisms of death pathway genes FAS and FASL and risk of premalignant gastric lesions. Anticancer Res 2008; 28: 97–103.
Gormus U, Ergen A, Yilmaz H, Dalan B, Berkman S, Isbir T : Fas-1377A/G and FasL-844 T/C gene polymorphisms and epithelial ovarian cancer. Anticancer Res 2007; 27: 991–994.
Erdogan M, Karadeniz M, Berdeli A et al: Fas/Fas ligand gene polymorphism in patients with papillary thyroid cancer in the Turkish population. J Endocrinol Invest 2007; 30: 411–416.
Yang M, Sun T, Wang L et al: Functional variants in cell death pathway genes and risk of pancreatic cancer. Clin Cancer Res 2008; 14: 3230–3236.
Qiu D, Kurosawa M, Lin Y et al: Overview of the epidemiology of pancreatic cancer focusing on the JACC Study. J Epidemiol 2005; 15 (Suppl 2): S157–S167.
Wei Q, Cheng L, Amos CI et al: Repair of tobacco carcinogen-induced DNA adducts and lung cancer risk: a molecular epidemiologic study. J Natl Cancer Inst 2000; 92: 1764–1772.
Cohen SM, Shirai T, Steineck G : Epidemiology and etiology of premalignant and malignant urothelial changes. Scand J Urol Nephrol Suppl 2000; 205: 105–115.
Blot WJ, McLaughlin JK, Winn DM et al: Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res 1988; 48: 3282–3287.
Au WW, Abdou-Salama S, Sierra-Torres CH, Al-Hendy A : Environmental risk factors for prevention and molecular intervention of cervical cancer. Int J Hyg Environ Health 2007; 210: 671–678.
Persson I : Estrogens in the causation of breast, endometrial and ovarian cancers – evidence and hypotheses from epidemiological findings. J Steroid Biochem Mol Biol 2000; 74: 357–364.
Mantel N, Haenszel W : Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959; 22: 719–748.
DerSimonian R, Laird N : Meta-analysis in clinical trials. Control Clin Trials 1986; 7: 177–188.
Whitehead A, Whitehead J : A general parametric approach to the meta-analysis of randomized clinical trials. Stat Med 1991; 10: 1665–1677.
Egger M, Davey Smith G, Schneider M, Minder C : Bias in meta-analysis detected by a simple, graphical test. BMJ 1997; 315: 629–634.
Thakkinstian A, McElduff P, D’Este C, Duffy D, Attia J : A method for meta-analysis of molecular association studies. Stat Med 2005; 24: 1291–1306.
Kabelitz D, Pohl T, Pechhold K : Activation-induced cell death (apoptosis) of mature peripheral T lymphocytes. Immunol Today 1993; 14: 338–339.
DeBruin LS, Josephy PD : Perspectives on the chemical etiology of breast cancer. Environ Health Perspect 2002; 110 (Suppl 1): 119–128.
Suzuki N, Wakisaka S, Takeba Y, Mihara S, Sakane T : Effects of cigarette smoking on Fas/Fas ligand expression of human lymphocytes. Cell Immunol 1999; 192: 48–53.
Elnemr A, Ohta T, Yachie A et al: Human pancreatic cancer cells express non-functional Fas receptors and counterattack lymphocytes by expressing Fas ligand; a potential mechanism for immune escape. Int J Oncol 2001; 18: 33–39.
Kelsey JL, Gammon MD : The epidemiology of breast cancer. CA Cancer J Clin 1991; 41: 146–165.
Song RX, Santen RJ : Apoptotic action of estrogen. Apoptosis 2003; 8: 55–60.
Akhmedkhanov A, Zeleniuch-Jacquotte A, Toniolo P : Role of exogenous and endogenous hormones in endometrial cancer: review of the evidence and research perspectives. Ann NY Acad Sci 2001; 943: 296–315.
Mor G, Kohen F, Garcia-Velasco J et al: Regulation of fas ligand expression in breast cancer cells by estrogen: functional differences between estradiol and tamoxifen. J Steroid Biochem Mol Biol 2000; 73: 185–194.
Dong L, Wang W, Wang F et al: Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J Biol Chem 1999; 274: 32099–32107.
Acknowledgements
We thank Qingyi Wei (Department of Epidemiology, The University of Texas MD Anderson Cancer Center) for critical comments and scientific editing and Guangfu Jin for his assistance in data analysis.
This study was partly supported by the National Natural Science Foundation of China (30271105, 30571583, 30800926, and 30872084), the PhD Programs Foundation of Ministry of Education of China (20060312002), the Natural Science Foundation of Jiangsu Province (BK2006231), the Postdoctoral Science Foundation of China (20060390293), the Postdoctoral Science Foundation of Jiangsu Province (0601049) and ‘Qinglan Project’ Foundation for the Young Academic Leader of Jiangsu Province (2006).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zhang, Z., Qiu, L., Wang, M. et al. The FAS ligand promoter polymorphism, rs763110 (−844C>T), contributes to cancer susceptibility: evidence from 19 case–control studies. Eur J Hum Genet 17, 1294–1303 (2009). https://doi.org/10.1038/ejhg.2009.45
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.45
Keywords
This article is cited by
-
Susceptibility to oral cancers with CD95 and CD95L promoter SNPs may vary with the site and gender
Tumor Biology (2015)
-
FasL Gene -844T/C Mutation of Esophageal Cancer in South China and Its Clinical Significance
Scientific Reports (2014)
-
Associations between Fas/FasL polymorphisms and susceptibility to cervical cancer: a meta-analysis
Tumor Biology (2014)
-
Association between CD95L polymorphism and cervical cancer risk: evidence from a meta-analysis
Tumor Biology (2014)
-
CD95 rs1800682 polymorphism and cervical cancer risk: evidence from a meta-analysis
Tumor Biology (2014)
