- NEWS AND VIEWS
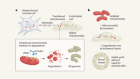
Actin proteins assemble to protect the genome
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 559, 35-37 (2018)
doi: https://doi.org/10.1038/d41586-018-05339-y
References
de Lanerolle, P. & Serebryannyy, L. Nature Cell Biol. 13, 1282–1288 (2011).
Belin, B. J. & Mullins, R. D. Nucleus 4, 291–297 (2013).
Caridi, C. P. et al. Nature 559, 54–60 (2018).
Schrank, B. R. et al. Nature 559, 61–66 (2018).
Chiolo, I. et al. Cell 144, 732–744 (2011).
Tsouroula, K. et al. Mol. Cell 63, 293–305 (2016).
Jakob, B. et al. Nucleic Acids Res. 39, 6489–6499 (2011).
Belin, B. J., Lee, T. & Mullins, R. D. eLife 4, e07735 (2015).
Andrin, C. et al. Nucleus 3, 384-395 (2012).
Chuang, C. H. et al. Curr. Biol. 16, 825–831 (2006).
Dundr, M. et al. J. Cell Biol. 179, 1095–1103 (2007).
Cho, N. W., Dilley, R. L., Lampson, M. A. & Greenberg, R. A. Cell 159, 108–121 (2014).
Aymard, F. et al. Nature Struct. Mol. Biol. 4, 366–374 (2014)
Massaad, M. J., Ramesh, N. & Geha, R. S. Ann. NY Acad. Sci. 1285, 26–43 (2013).
 Read the paper: Nuclear F-actin and myosins drive relocalization of heterochromatic breaks
Read the paper: Nuclear F-actin and myosins drive relocalization of heterochromatic breaks
 Read the paper: Nuclear ARP2/3 drives DNA break clustering for homology-directed repair
Read the paper: Nuclear ARP2/3 drives DNA break clustering for homology-directed repair
 Actin filaments up against a wall
Actin filaments up against a wall
 Actin’ dangerously
Actin’ dangerously