
Abstract
Three T-DNA insertional embryonic lethal mutants from NASC (The Nottingham Arabidopsis Stock Center) were first checked with their segregation ratio of abortive and normal seeds and the copy number of T-DNA insertion. The N4081 mutant has a segregation ratio of 1:3.04 in average and one T-DNA insertion site according to our assay. It was therefore chosen for further analysis.
To isolate the joint fragment of T-DNA and plant DNA, the plasmid rescue technique was used. pEL-7, one of plasmids from left border of T-DNA, which contained pBR322 was selected from ampicillin plate. The T-DNA fragment of pEL-7 was checked by restriction enzyme analysis and Southern Blot. Restriction analysis confirmed the presence of known sites of EcoRI, PstI and PvuII on it. For confirming the presence of flanking plant DNA in this plasmid, pEL-7 DNA was labeled and hybridized with wild type and mutant plant DNA. The Southern Blot indicated the hybridization band in both of them.
Furthermore, the junction of T-DNA/plant DNA was subcloned into bluescript SK+ and sequenced by Applied Biosystem 373A Sequencer. The results showed the 822 bp fragment contained a 274 bp sequence, which is 99.6% homolog (273bp/274 bp) to Ti plasmid pTi 15955 DNA. Ten bp of left 25 bp border repeat were also found in the juction of T-DNA and Plant DNA.
Taken together, pEL-7 should contain a joint fragment of T-DNA and flanking plant DNA. This plasmid DNA could be used for the isolation of plant gene, which will be helpful to elucidate the relationship between gene function and plant embryo development.
Similar content being viewed by others


Introduction
Embryogenesis is one of the key event of plant development, in which a series of important processes, such as body plan and cell differentiation pattern, were involved. For many years, the attention was paid on comparative morphology and cell division patterns (Meinke 1991)1. There was only limited knowledge of its biochemical and molecular aspects because of small size of embryo, in spite of the fact that considerable progress has been achieved in Drosophila by using mutants. Later, Arobidopsis embryonic lethal mutant was used as a model system for plant embryogenesis investigations (Meinke 1979)2. The embryonic mutants can be induced by x-irradiation (Mller 1963)3, chemical mutagens (Meinke 1985, Jurgens et al 1991)4, 5, transposable elements (Clark and Sheridan 1991)6 and T-DNA insertion by Agrobacterium (Koncz et al 1989, Errampalli et al 1991)7, 8. Therefore, mutant emlyos will provide a unique opportunity for further investigation on plant embryo development.
Embryonic lethal or embryonic defective mutants as named recently (Meinke 1995)9 are commonly found after above treatments. The mutant embryo usually has partialy defect in embryo and was arrested at a given developmental phase. Therefore, the lethal mutant would facilitate analyzing the genes that perform essential function in different stages of embryogenesis. One of the problems of embryonic lethal analysis is that homozygous mutant individuals often die before maturity. For this approach, one of the effective techniques is plasmid rescue. By this method, the joint fragment of inserted T-DNA tagging sequence and the flanking plant DNA can be recovered by plasmid rescue technique from heterozygous T-DNA insertional mutant plant. The flanking sequence can then be served a as a starting point for the isolation of plant gene. Recently, the plasmid rescue technique has been applied to the investigation of plant development such as flower development (Yanofsky et al 1990) 10. In the present paper, we use this technique to recover the joint fragment of T-DNA and flanking plant DNA sequence from T-DNA inserted embryonic lethal mutant of Arabidopsis thaliana.
Materials and Methods
Plant materials
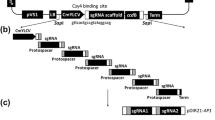
The wild-type seed of Arabidopsis thaliana (Columbia) and N4160, N4081, N4296 embryonic lethal mutants seeds were kindly provided by NASC (The Nottingham Arabidopsis Stock Center), which was donated by Dr. Csaba Koncz, Max-Planck-Institut. N4160 mutant was induced by pPCV621 T-DNA insertion. N4081, N4296 mutants were induced by pPCV6NFHyg T-DNA insertion (Fig 1).

Schematic representation of the gene tagging vector pPCV6NFHyg and cloning of the disrupted plant DNA by plasmid rescue. The vector harbors a plasmid rescue cassette with antibiotic resistance marker (Apr , Cbr) and a host origin of replication (pBR322) and other bacterial function. Apr, Ampicillin resistance; Cbr, Carbenicillin resistance; Aps, Ampicillin sensitiveness; ocs, octopine synthase; nos, nopaline synthase; aph(3')II, aminoglycoside phosphotransferase; HPH, hygromycin resistant gene; A4, ipt gene; g5, TL-DNA gene 5; TL-DNA, T-DNA from Ti plasmid Ach5. LB represents T-DNA left border repeat. RB represents T-DNA right border repeat. The data of pPCV6NFHyg was from personal communication of Dr. Csaba Knocz.
Plant growth conditions
Seeds were sown on wet mixture of soil and vermiculite in plastic pots. The pots were placed in 4°C for 4 d to break dormancy and then exposed to 22°C under 16 h light / 8 h dark cycles. The seedlings were watered from bottom.
Plant tissue culture
For producing callus, leaves from six-week-old seedlings of Arabidopsis thaliana were sterilized with 70% ethanol and rinsed with distilled water several times. The axenic leaves were cut into small segments and transferred to 25 ml solidified MS medium (Murashage 1962)11 supplemented with 2.0 mg/1 2, 4D. Then they were incubated at 22°C under a 16 h light / 8 h dark cycle. The callus from different mutants and wild type of Arabidopsis thaliana were used for further DNA isolation.
Determination of the copy number of T-DNA insertional mutants of N4160, N4081, N4296
The copy number was determined as discribed by Maniatis 198212 with modification. DNA was prepared from callus of different mutants according to James et al 199213 and digested with EcoRI (2-3 μg DNA per reaction), electrophoresed, blotted onto nitrocellulose membrane, and hybridized with non-radioactive Digoxigenin(Dig)-laheled pBR328 probe. Hybridization was performed in 5×SSC, 1% blocking reagent (w/v), 0.1% N-lauroylsarcosine, Na-salt (w/v), 0.02% SDS (w/v) at 68°C. DNA gel blots were visualized by immunological detection methods, using Boehringer Mannheim Kit (see DNA labeling and Southern hybridization below).
Plasmid rescue
The procedure of plasmid rescue is mainly according to Behringer and Medford 199214 with modification, the electroporation was not used in this paper. The main steps of plasmid rescue were summarized in Fig 1.
DNA was isolated from N4081 mutant callus that contained a single copy of integrated T-DNA. 2μg DNA was digested with EcoRI in 20μl volume. Incubate at 37°C for 2 h. After digestion with EcoRI and phenol extraction, add 1/10 volume of 3.0 M sodium acetate and 2 volumes of 100% ethanol and the DNA was precipitated at -20°C for 1 h. The DNA was resnspended in 15μl TE buffer (10 mM Tris, l mM EDTA, pH7.0). Add 2μl 10 ms M ATP, 2μl l0×ligation buffer and 1 μl (8U) T4 DNA ligase (Boehringer Mannheim) to the EcoRI-digested DNA. Incubate at 16°C for 24 h. Self-ligated fragments were transformed into competent cells of TG1 by the standard CaC12 transformation procedure (Sambrook et al 1989)15.
Screening of ampicillin resistant colonies
Plasmid DNA was prepared from individual colonies using a standard alkaline lysis miniprep DNA procedure (Sambrook et al 1989)15. The miniprep DNA was digested with EcoRI, PstI, PvuII in different combination or respectively. Then the digests were electrophoresed on a 1.0% agarose gel in lx TAE running buffer.
DNA labeling and southern hybridization
Plasmid DNA labeling
In the present paper, DNA was labeled by Digoxigenin(Dig) using Dig DNA labeling and detection Kit (Boehringer Mannheim).
For probe purification, 2μg linearized DNA was purified by phenol / chloroform extraction, ethanol precipitation, and resuspended in 15μl distilled water. The DNA was denatured by heating in a boiling water for 10 min and chilling quickly on ice / NaC1. Complete denaturation is essential for efficient labeling.
Southern blot
Southern Blot was conducted by using Pharmacia LKB VacuGene apparatus. The wild type Arabidopsis total DNA and N4081 mutant total DNA were digested with EcoRI, electrophoresed on a 1.0% agarose gel, and then blotted onto nitrocellulose membrane, and hybridized by Dig-labeled plasmid DNA probe. Hybridization was performed at 68 °C. DNA gel blots were visualized according to the procedure of Boehringer Mannheim Kit mentioned above.
Sequence analysis of joint fragment of T-DNA and plant DNA
The junction region of the T-DNA/plant DNA was isolated from pEL-7 through ClaI and XbaI digestion, agarose electrophoresis and then subcloned into bluescript SK. The sequence was analyzed by Applied Biosystem 373A Sequencer.
Results
Segregation ratios in N4081 heterozygous siliques
All seeds are identical in colour (milky white or colourless) and size within 3 d after flowering. So it is difficult to distinguish aborted seeds from normal seeds. But aborted seeds of N4081 mutant can be unambiguously identified by their size or colour 9 d after flowering under dissecting microscope. At this stage, normal seeds are green and have reached mature size, while aborted seeds are smaller and in milky white, colourless, or begin to degenerate and turn brown. Ten more siliques were tested for the presence of aborted seeds. The result indicated that the N4081 heterozygous siliques contained approximately 25% aborted seeds following self-pollination. Their segregation ratio of aborted seeds and normal seeds is 1:3.04 in average. In the meantime, the hygromycin resistant was also assayed, the results observed after 3 weeks germination indicated a ratio of 2.2:1 for hyg(r):hyg(s). So N4081 mutant appears to be segregated for a single recessive gene.
Determination of the copy number of T-DNA insertions
Using Dig labeled pBR328 probe, the copy number of T-DNA insertional mutants was estimated. The results showed that both N4160 and N4081 mutants contain one integrated T-DNA copy, but N4296 mutant contains two T-DNA copies (Fig 2). Taken together with the results of segregation, this confirmed that N4081 mutant was induced by a single T-DNA integration. Therefore, N4081 mutant was selected for further studies by plasmid rescue.

Determination of the Copy Number of T-DNA insertions. After digestion with EcoRI, the DNA was resolved on a 1.0% agarose gel, then blotted onto a nitrocellulose membrane, and hybridized by Dig-labeled pBR328, lane 1: wild-type Arabidopsis total DNA; lane 2:N4160 mutant total DNA; lane 3: N4081 mutant total DNA; lane 4: N4296 mutant total DNA.
Plasmid rescue
The vector of pPCV6NFHyg in N4081 mutant contained a fragment of pBR322. Therefore, it facilitates to clone the joint fragment of T-DNA and plant flanking DNA by plasmid rescue. For approaching that, first of all, we optimized the condition of self-ligation and transformation.
DNA concentration is important for self-ligation. N4081 mutant total DNA was digested with EcoRI and different concentrations of DNA were used in self-ligation reaction in order to characterize the optimal range of DNA concentration. The results indicated that DNA concentration below 50 ng / μl is appropriate. The higher DNA concentration usually gave a low self-ligation efficiency.
To improve the frequency of transformation, four different E.coli strains, K802, JM83, DH5α and TG1 were selected by the standard CaC12 transformation procedure. Compared with other strains, TG1 has a higher efficiency of transformation. Under suitable conditions, a transformation frequency, which is comparable to the results of Beringer and Medford14, could be obtained. The clones are resistant to ampicillin and have a negative response in X-gal dish.
Characterization of T-DNA insertion in pEL-7 plasmid
After digestion with EcoRI, pEL-7 plasmid DNA and N4081 total DNA were resolved on a 1.0% agarose gel, blotted onto nitrocellulose membrane, hybridized with Dig-labeled pBR328 probe. It was found both EcoRI digested N4081 mutant DNA and pEL-7 plasmid DNA have the same band of ∼ 6Kb. Accordingly, the rescued joint fragment of T-DNA and flanking plant DNA is identical to those from N4081 mutant (Fig 3).

Southern hybridization of the pEL-7 plasmid. Both pEL-7 plasmid DNA and N4081 mutant total DNA were digested with EcoRI, electrophoresed on a 1.0% agarose gel, blotted onto nitrocellulose membrane, and hybridized with Dig-labeled pBR328 probe, lane 1: pEL-7 plasmid DNA; lane 2:N4081 mutant total DNA.
Furthermore, the restriction enzyme analysis also confirmed the presence of known sites of EcoRI, PstI and PvuII in the T-DNA part of pEL-7. When pEL-7 was digested by EcoRI / PstI; EcoRI / PvuII; PvuII and EcoRI, the ∼700bp, ∼5.3Kb (Fig 4A); ∼2.2Kb, ∼2.3Kb, ∼l.5Kb; ∼2.2Kb, ∼3.8Kb; ∼6Kb bands (Fig 4B) appeared respectively.

Characterization of pEL-7 plasmid by restriction analysis and Southern Blot hybridization.
A: lane 1: Hind III-restricted lambda DNA marker; lane 2: EcoRI / PstI digested pEL-7 plasmid DNA. B:lane l: Hind III-restricted lambda DNA marker; lane 2: plasmid EcoRI / PvuII digested pEL-7 plasmid DNA. lane 3: PvuII digested pEL-7 plasmid DNA. Land 4: EcoRI digested pEL-7 plasmid DNA. C: southern Blot hybridization of the gel shown in B, using the Dig-labeled PBR328 as the probe.
The further Southern Blot indicated, when pEL-7 was digested with EcoRI, PvuII, EcoRI / PvuII respectively. There are only ∼6Kb; ∼3.8Kb; ∼2.3Kb bands hybridized (Fig 4C). It was shown that these bands contained pBR322 fragment of T-DNA and other unhybridized bands were likely the fragment of plant DNA.
Characterization of the flanking plant DNA in pEL-7 plasmid
Using Dig labeled plasmid DNA probe, the Dot Blot hybridization indicated that the flanking plant DNA in this plasmid is homologous to wild type Arabidopsis DNA. Furthermore, we transferred EcoRI digested wild type Arabidopsis total DNA and N4081 mutant total DNA to nitrocellulose membrane and hybridized by Dig-labeled plasmid DNA probe. The results showed that both of them had the same band of ∼ 3Kb (Fig 5). Therefore, it confirmed that the rescued DNA should contain a fragment of flanking plant DNA. The N4081 mutant total DNA had another band of ∼6Kb (Fig 5). This is due to the presence of T-DNA and plant DNA joint fragment in heterozygous plant. The sequencing of this flanking plant DNA is under processing.

Characterization of the flanking plant DNA in pEL-7 plasmid. After digested with EcoRI, N4081 total DNA and wild-type Arabidopsis total DNA were resolved on a 1.0% agarose gel, blotted onto nitrocellulose membrane, hybridized with Dig-labeled pEL-7 plasmid probe. Lane l: N4081 total DNA; lane 2: wild-type Arabidopsis total DNA.
Cloning and sequencing of T-DNA /plant DNA junction
For subcloning the junction region of T-DNA and plant DNA, the restriction enzyme assay was used to find the suitable sites in plant DNA part from pEL-7 DNA.Then the junction fragment of T-DNA / plant DNA between ClaI and XbaI sites was subcloned into bluescript SK+. The sequence of this clone was further analyzed by Applied Biosystem 373A Sequencer. By means of BLASTN searching in genbank, the results showed that this fragment includes two parts. In the first part of sequence, from 1-274 bp was 99.6% (273bp / 274bp) homolog to Ti plasmid pTi 15955 and 10 bp of left 25 bp border repeat was 90.9% homolog with left border repeat (10bp / 11bp) and located from 275 to 284bp, where is just at the junction between this two parts.This part of sequence is clearly from T-DNA vector. The second part of sequence from 285 to 822 bp is plant DNA, the putative TATA box was found from 453 to 460 bp, according to computing analysis (Fig 6).

The sequence of T-DNA / plant DNA junction from pEL-7
From 1-274 is T-DNA fragment (99.6% homolog to Ti plasmid DNA); from 275-284 is the left border repeat; from 285-822 is the plant DNA; from 453-460 is putative TATA signal. EMBL accession#:Y09563.
Discussion
The objective of this paper is to isolate the flanking plant DNA sequence from T-DNA inserted mutant in order to study the function of the tagged gene in embryo development. To confirm the relationship between embryonic lethal and T-DNA insertion, the phenotype of embryonic lethal mutant (N4081) was identified by observation under dissecting microscope. More than 10 siliques were examined. The mutant embryo was arrested proximatly before the early stage of cotyledon (The identification of exact lethal phase is in progress). The segregation ratio of aborted seeds and normal seeds is 1:3.04 in average. In the meantime, the hygromycin resistant was also assayed. The results indicated a ratio of 2.2:1 for hyg (r): hyg(s). Furthermore, the T-DNA copy number was assayed by Southern Blot, When EcoRI digested transgenic plant DNA was hybridized with Dig-labeled pBR328 plasmid DNA, a single hybridized band indicated that N4081 mutant possessed only one T-DNA insertional site (Fig 2). Therefore, the N4081 mutant was chosen for further investigstion.
To isolate the adjacent fragment of T-DNA and flanking plant DNA, the plasmid rescue technique was applied in this paper. The first problem we have met is the relative low frequency transformation of E.coli. In our previous work, when higher concentration of EcoRI digested transgenic plant DNA was used for self-ligation, the frequency of transformation was very low. Sometimes, even no colony could be found in ampicillin plate. To improve the frequency of transformation, we optimized the condition of self-ligation and selected suitable host strain for transformation. Therefore, a transformation frequency which is comparable to the results of Beringer and Medford 199214 was obtained.
The rearrangement of T-DNA and plant DNA joint fragment is another problem for plasmid rescue. The rearrangement has been reported in rescued plant DNA (Ott and Chua 1990, Behringer and Medford 1992)16, 14 and both plant DNA and T-DNA. Some E.coli strains, such as DH5α, could cause rearrangement, due to their ability to restrict C-methylated DNA (Mandal 1993)17. To eliminate rearrangement, one E.coli strain TG1 that neither modify nor restrict transformed DNA was used in our work.
The rescued colonies were screened according to the molecular weight of EcoRI digested LB border of T-DNA and then checked by restriction enzyme analysis and Southern Blot. pEL-7 is one of the selected colony with molecular weight of a.bout 6Kb (Fig 3). The restriction enzyme analysis confirmed the presence of known restriction sites (EcoRI. PstI. PvuII) in T-DNA. When it was hybridized by Diglabeled pBR328 plasmid DNA, the hybridized band of EcoRI digested N4081 mutant DNA and pEL-7 DNA is identical, which indicated that pEL-7 plasmid DNA contained T-DNA tagging. The pEL-7 was further labeled to hybridize with wild type Arabidopsis DNA. The results showed that one identical band appeared in both EcoRI digested wild type and N4081 mutant Arabidopsis DNA. Finally, the junction of T-DNA and plant DNA was subcloned into Bluescript SK+. The sequence analysis was further confirmed the presence of 274 bp fragmnet which was 99.6% homolog to Ti plasmid DNA and 10 bp of left 25 bp border repeat at the junction between T-DNA and plant DNA. The part of plant DNA contained a putative TATA box (from 453 to 460 bp) and a short exon fragment according to computing analysis. Since the plant DNA fragment may contain some deletion or rearangement ,the plant gene disrupted by T-DNA needs to be further characterized by screening of cDNA liberary. Accordingly, the rescued colony pEL-7 contained both T-DNA and flanking plant DNA. This joint fragment of T-DNA and plant DNA therefore could be served as a starting point for further cloning of plant sequence that regulates embryo development.
References
Meinke DW . Perspectives on genetic analysis of plant embryogenesis. Plant Cell 1991; 3:857–66.
Meinke DW, Sussex IM . Embryo-lethal mutants of Arabidopsis thaliana, A model system for genetic analysis of plant development. Dev Biol 1979; 72: 50–61.
Muller AJ, Embryonentest zum nachweis rezessiver letafaktoren bei Arabidopsis thaliana. Biol zentralbl 1963; 82:133–63.
Meinke DW . Embryo-lethal mutants of Arabidopsis thaliana: Analysis of mutants with a wide range of lethal phase. Theor Appl Genet 1985; 69:543–52.
Jurgens G, Mayer U, Torres RA, Berleth T, Misera S . Genetic analysis of pattern formation in the Arabidopsis embryo. In Molecular and cellular basis of pattern formation. K, Roberts. Cambridge, company of Biologists, Ltd. 1991; pp27–38
Clark JK, Sheridan WF . Isolation and characterization of 51 embryo-specific mutations of maize. Plant Cell 1991; 3:935–51.
Koncz C, Martini N, et al. High-frequency T-DNA mediated gene tagging in plants. Proc Natl Acad Sci USA 1989; 86:8467–71.
Errampalli D, Patton D, Castle L, Mickelson L, Hanse K, et al. Embryonic lethal and T-DNA insertional mutagenesis in Arabidopsis. Plant Cell 1991; 3:149–57.
Meinke DW . Molecular genetics of plant embryogenesis. Ann Rev Plant Physiol Plant Mol Biol 1995; 46:369–94.
Yanofsky MF, Ma H, Bowman JL, et al. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 1990; 346:35–9.
Murashage T, Skoog F . A revised medium for rapid growth and bioassays with tobacco tissue cultures. Plant Physiol 1962; 15:473–97.
Maniatis T, Fritsch EF, Sambrook J . Molecular cloning: a laboratory manual (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) 1982.
James HO, Sridhar D . Rapid isolation of rice and maize DNA for analysis by random-primer PCR. Plant Molecular Biology Reporter 1992; 10(3):236–41.
Behringer FJ, Medford JI . A plasmid rescue technique for the recovery of plant DNA disrupted by T-DNA insertion. Plant Molecular Biology Reporter 1992; 10(2):190–8.
Sambrook J, et al. Molecular cloning: a laboratory manual, 2nd edition, Cold Spring Harbor Laboratory Press 1989.
Ott RW, Chua NH . Enhancer sequences from Arabidopsis thaliana obtained by library trans- formation of Nicotiana tabacum. Mol Gen Gent 1990; 223:169–79.
Mandal A, Lang V, Orczyk W, Palva ET . Improved efficiency for T-DNA-mediated transfor- mation and plasmid rescue on Arabidopsis thaliana. Theor Appl Genet 1993; 86:621–8.
Acknowledgements
The authors want to express thanks to Dr. Csaba Koncz, Max-Plank-Institut and NASC (The Nottingham Arabidopsis Stock Centre) for providing mutant seeds.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Yao, X., Sun, J. & Zhu, Z. Isolation of T-DNA flanking plant DNA from T-DNA insertional embryo-lethal mutants of Arabidopsis thaliana by plasmid rescue technique. Cell Res 6, 125–136 (1996). https://doi.org/10.1038/cr.1996.14
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/cr.1996.14
