
Abstract
Age-related macular degeneration (AMD) is a degenerative disease of the retina and the leading cause of blindness in the elderly in developed countries. The late stage of dry AMD, or geographic atrophy (GA), is characterized by extensive retinal pigment epithelium (RPE) degeneration. The underlying molecular mechanism for RPE cell death in GA remains unclear. Our previous study has established that RPE cells die predominantly from necroptosis in response to oxidative stress in vitro. Here, we extend our study and aim to characterize the nature of RPE cell death in response to sodium iodate (NaIO3) in vitro and in a NaIO3-induced retina degeneration mouse model. We found that NaIO3 induces RPE necroptosis in vitro by using a combination of molecular hallmarks. By using TUNEL assays, active caspase-3 and HMGB1 immunostaining, we confirmed that photoreceptor cells die mainly from apoptosis and RPE cells die mainly from necroptosis in response to NaIO3 in vivo. RPE necroptosis in this model is also supported by use of the RIPK1 inhibitor, Necrostatin-1. Furthermore, using novel RIPK3-GFP transgenic mouse lines, we detected RIPK3 aggregation, a hallmark of necroptosis, in the RPE cells in vivo after NaIO3 injection. Our findings suggest the necessity of re-evaluating RPE cell death mechanism in AMD models and have the potential to influence therapeutic development for dry AMD, especially GA.
Similar content being viewed by others


Age-related macular degeneration (AMD) is a degenerative disorder of the macula and the leading cause of irreversible central vision loss in the elderly population in the developed countries.1 The dry form of AMD is characterized by a yellow deposit called ‘drusen’ under the retina at the early stage and geographic atrophy (GA) at the late stage. GA is manifested in scattered or confluent areas of degeneration of retina pigment epithelial (RPE) cells. RPE degeneration is thought to result in the degeneration of the overlying photoreceptors and eventually vision loss.
Age is the most consistent risk factor associated with AMD. Genetic factors, oxidative stress, inflammation, and ethnicity are considered to be contributors to the pathogenesis of AMD.2 Among them, oxidative stress has been suggested as a critical component of AMD pathogenesis.3 Cigarette smoking, which induces systemic oxidative stress, has been demonstrated to be a significant risk factor for AMD. Clinical studies have shown that the progression of AMD can be slowed with antioxidant vitamins and zinc supplements.4,5 The complete pathological mechanism underlying dry AMD has not been completely understood, and the disease is currently untreatable.
Sodium iodate (NaIO3) injection has been extensively used as a pre-clinical model of RPE dystrophy and GA.6 NaIO3-induced retinal degeneration displays two features similar to AMD. First, low doses lead to a patchy loss of the RPE cells leaving spots void of autofluorescence as in GA. Second, the RPE loss not only affects the photoreceptors but also the underlying choriocapillaris.7 NaIO3 is thought to directly affect the RPE cells with secondary effects on photoreceptors and the choriocapillaris and has been shown to induce the production of reactive oxygen species contributing to damages in RPE cells.8,9 Other effects of NaIO3 on RPE cells include: inhibition of enzyme activity (e.g., triose phosphate dehydrogenase, lactate dehydrogenase) in RPE cells, disruption of the blood–retina barrier, and increased conversion of glycine to potentially toxic glyoxylate by melanin.10–12
Two major types of cell death, apoptosis and necroptosis, occur in response to oxidative stress.13 Apoptosis is characterized by maintenance of the plasma membrane, chromatin condensation and fragmentation, and caspase activation. Necroptosis is a regulated form of necrosis mediated by receptor-interacting protein kinases (RIPK).14 In contrast to apoptosis, necroptosis is characterized by ATP depletion, rupture of the plasma membrane, and release of necroptosis-specific cytokine HMGB1 to activate inflammatory response.15,16 Owing to the different implications in inflammatory response between apoptosis and necroptosis, to develop targeted therapy for AMD, it is crucial to clarify the mechanism of RPE cell death in response to oxidative stress and in AMD. We recently found that the molecular features of apoptosis were not observed in RPE cells in response to H2O2 or tBHP treatment.17 Instead, cardinal features of necroptosis, including ATP depletion, RIPK3 aggregation, and the release of HMGB1 from the nucleus were detected. Inhibition of RIPK activity by necrostatins or downregulation of RIPK3 by siRNAs largely rescued oxidative stress-induced RPE death. Our results suggest that RPE necroptosis is the predominant mechanism of RPE cell death in response to oxidative stress in vitro.
Here, we extend our previous study and characterize the nature of RPE cell death in both in vitro and in vivo NaIO3 models. We provide evidence that NaIO3 induces RPE necroptosis, but not apoptosis in vitro. We also utilize a NaIO3-induced RPE degeneration model to analyze the molecular changes associated with RPE and photoreceptor cell death in vivo, and confirm photoreceptor apoptosis and RPE necroptosis in response to NaIO3 in vivo. RPE necroptosis in the model is supported by use of RIPK1 inhibitor, Necrostatin-1. By using novel RIPK3-GFP transgenic mouse lines, we detected RIPK3 aggregation, a hallmark of necroptosis, in the RPE cells after retro-orbital NaIO3 injection, further confirming RPE necroptosis in vivo.
Results
Sodium iodate-induced RPE necroptosis in vitro
The NaIO3 animal model has been widely used as a pre-clinical model of RPE dystrophy and atrophic AMD. Surprisingly, very few studies documented the effect of NaIO3 in RPE cells in vitro.18,19 We first determined the half maximal effective concentration (EC50) of NaIO3 in confluent ARPE-19 cells. After testing the RPE cell survival at 24 h after different concentrations of NaIO3 (0–75 mM) treatment, the EC50 of NaIO3 in ARPE-19 cells was calculated to be 10.5 mM (Supplementary Figure 1); 10 mM NaIO3 was then selected for the subsequent in vitro experiments.
To examine the nature of NaIO3-induced RPE cell death in vitro, we tested cell membrane permeability to propidium iodide (PI), an established hallmark of necrosis. Confluent ARPE-19 cells were subjected to DAPI/PI staining at 24 h after treatment with 10 mM of NaIO3. Strong cytoplasmic and nuclear staining with PI was observed in the treated but not the control cells (Figure 1A (a and b)). We have previously shown that necrotic ARPE-19 cells show RIPK3 aggregation and HMGB1 nuclear release.17 We analyzed the cellular distribution of RIPK3 by transfecting ARPE-19 cells with an RIPK3-GFP-expressing plasmid. RIPK3 was evenly distributed in the cytoplasm under normal condition, but formed punctuates in the periphery region of cells within 2 h of 10 mM NaIO3 treatment, indicating necrosome formation (Figure 1A (c and d)). HMGB1 distribution and mitochondria morphology were visualized in ARPE-19 cells after co-transfecting with plasmids expressing HMGB1-YFP and ANT1-RFP. HMGB1 is a chromatin structural protein detected inside the nucleus. Within 4 h of NaIO3 treatment, HMGB1 was released to the cytoplasm as indicated by the distribution of HMGB1-YFP signal (Figure 1A (e and f)). The mitochondria form a tubular network as visualized after ANT1-RFP transfection. At 4 h of NaIO3 treatment, mitochondrial network was fragmented and clustered in the perinuclear region (Figure 1A (e and f)). Taken together, these results suggest that necroptosis but not apoptosis mainly accounts for cell death in ARPE-19 cells in response to NaIO3 treatment.

Evidence for sodium iodate-induced RPE necroptosis in vitro. (A) NaIO3 (10 mM) treatment induces changes to cell membrane permeability in non-fixed ARPE-19 cells shown by staining with PI (a and b). Activation of RIPK3 after 2 h of 10 mM NaIO3 treatment in RIPK3-GFP-transfected ARPE-19 cells (c and d). Mitochondrial network was tracked by ANT1-RFP and passive release of HMGB1 from the nucleus by HMGB1-YFP at 4 h after NaIO3 treatment (e and f). (B) ARPE-19 cells were treated with 200 μM Nec-1, -5, -7; 3 μM GSK’872, or 25 μM resveratrol for 24 h before 10 mM NaIO3 treatment. Cell viability was measured by MTT assay at 24 h. (C) ARPE-19 cells were treated with 50 μM z-VAD for 24 h before 10 mM NaIO3 treatment. Cell viability was measured by MTT assay at 24 h later. (D) Activation of inflammasome was tracked by transfection of ASC-GFP-expressing plasmid in ARPE-19 cells. Alu RNA (5 ng) was used as a positive control for inflammasome activation (a and b). Treatment with 10 mM NaIO3 did not induce inflammasome activation (c and d). (E) ARPE-19 cells were treated with 50 μM Ac-YVAD for 24 h before 10 mM NaIO3 treatment. Cell viability was measured by MTT assay at 24 h later. *P<0.05; **P<0.01; ***P<0.001. The scale bar is 25 μM.
To further confirm the necrotic nature of cell death in RPE cells in response to NaIO3 treatment, ARPE-19 cells were treated with different cell death inhibitors before exposure to NaIO3. We pretreated APRE-19 cells with 200 μM of Nec-1, -5, or -7 for 24 h and exposed them to 10 mM of NaIO3. As a positive control, resveratrol (25 μM) rescued up to 93% of ARPE-19 cells from NaIO3-induced death.19 Nec-1, a direct RIPK1 inhibitor, increased ARPE-19 cell survival from 47 to 75%. Nec-5, an indirect RIPK1 inhibitor, increased ARPE-19 cell survival to 67%, while Nec-7 that targets RIPK1-independent necrosis pathways had no effect on ARPE-19 viability. In addition, GSK’872, a specific RIPK3 inhibitor, protected up to 67% ARPE-19 cells from NaIO3-induced cell death (Figure 1B). However, z-VAD, a pan-caspase inhibitor, failed to protect ARPE-19 cells from NaIO3-induced death (Figure 1C). These observations indicate that NaIO3-induced RPE cell death is mainly necroptosis that depends on RIPK1 and RIPK3.
RIPK3 activation has been recently shown to be implicated in the induction of pyroptosis, a cell death pathway that relies on caspase-1 activation and inflammasome formation.20 To test whether NaIO3 can induce inflammasome activation in RPE cells, ARPE-19 cells were transfected with ASC-GFP, a component of the inflammasome.21 ASC-GFP-transfected ARPE-19 cells showed uniform cytoplasmic ASC-GFP expression. As a positive control, Alu RNA treatment induced inflammasome formation in ARPE-19 cells as shown by aggregated cytoplasmic ASC-GFP foci (Figure 1D (a and b)).22 When ARPE-19 cells were treated with NaIO3 for 0–24 h, aggregated ASC-GFP foci were not observed, indicating no inflammasome formation (Figure 1D (c and d)). To further confirm this observation, ARPE-19 cells were treated with 50 μM caspase-1 inhibitor Ac-YVAD for 24 h prior to NaIO3 treatment. No rescue of ARPE-19 cells was detected arguing against the involvement of pyroptosis in RPE cell death in response to NaIO3 (Figure 1E).
Taken together, our observations indicate that NaIO3 induces mainly RIPK1- and RIPK3-dependent necroptosis in ARPE-19 cells in vitro.
Morphological changes in the mouse retina after retro-orbital NaIO3 administration
Although NaIO3 administration has been widely used to induce RPE degeneration, there has been little in-depth analysis regarding the nature of RPE cell death in vivo in this model. NaIO3 has been shown to induce reactive oxygen species in RPE cells, making it an excellent model to study oxidative stress in vivo.23 We adopted a retro-orbital injection of low dose NaIO3 (20 mg/kg) to mouse retina to examine the nature of RPE cell death in vivo, with a focus to capture the early events leading to RPE death.24–26 H&E staining and methylene blue staining were used to validate the effect of NaIO3 administration in RPE cells. In control animals treated with saline, the RPE cell layer was evenly pigmented (Figure 2a and e) with RPE polarization noticeable in semi-thin plastic sections (Figure 2i). Furthermore, photoreceptor outer nuclear layer, inner segments, and outer segments were well organized. At 24 h after 20 mg/kg NaIO3 administration, RPE cells showed signs of loss of pigmentation with no visible effect on photoreceptor morphology (Figure 2b arrowhead, Figures 2f and j). At 48 h post injection, patchy RPE degeneration was observed: RPE appeared swollen, arrangement of photoreceptor segments was disorganized, and inner segments lost their regular organization (Figure 2c arrowhead, Figures 2g and k). At 72 h post injection, RPE cells appeared swollen, vacuolized, and started to break off from the RPE layer. Disorganization of outer nuclear layer, inner segments, and outer segment continued to progress reflecting photoreceptors cell death (Figure 2d arrowhead, Figures 2h and l). Throughout the time points, the RPE cell layer became progressively thinner. Taken together, our morphological analyses suggest necrotic death of RPE cells in the low-dose NaIO3 model, and validate this system for further molecular studies.

Histological staining of the mice retina after NaIO3 administration. (a–d) Hematoxylin and eosin staining of paraffin-embedded retinal cross sections of 20 mg/kg NaIO3-treated C57BL/6J mice, at different time points. At 24 h post administration, loss of RPE pigmentation was visible (arrowheads); RPE swelling and breaking of RPE monolayer was observed at 48 and 72 h, respectively (arrowheads). Scale bar is 25 μm. (e–h) Areas indicated by arrowheads at higher magnification. Scale bar is 10 μm. (i–l) Semi-thin sections and methylene blue staining revealed more details of the degenerated but still partially pigmented RPE cells with visible cell vacuolization at 72 h (arrowhead). Scale bar is 10 μm. ONL: photoreceptor outer nuclear layer, IS: inner segments, OS: outer segments. Results shown here are representative from six C57BL/6J mice at each time point.
Molecular characteristics of retinal cell death upon retro-orbital NaIO3 administration
To dissect the molecular changes in the retina after retro-orbital NaIO3 administration, tight junctions were visualized by immunostaining with antibody to ZO-1, a tight junction-associated protein. NaIO3 administration resulted in the loss of RPE monolayer properties as visualized by ZO-1 staining, at 48 h after NaIO3 injection, indicating a compromise of the blood–retina barrier (Figure 3a–d).

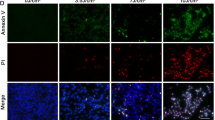
Molecular hallmarks of RPE degeneration in C57BL/6J mice RPE after retro-orbital NaIO3 administration. (a–d) Tight junctions in the central retina were visualized by ZO-1 staining. At 48 h post 20 mg/kg NaIO3 administration, compromise of tight junctions was visible indicating impairment of the blood–retina barrier (n=6 each). (e–h) Flatmount DAPI and PI staining highlighted necrotic RPE cells as early as 24 h post NaIO3 administration. At 72 h, areas with diffused PI signal were seen (n=6 each). (i–l) TUNEL staining was used to highlight dying cells in the retina. Both RPE cells and photoreceptors were TUNEL-positive as early as 24 h post 20 mg/kg NaIO3 administration (n=6 each). (m–p) Active caspase-3 staining was observed only in the photoreceptor layer at all analyzed time points (n=5 each). The scale bar is 25 μM.
Retro-orbital PI injection was further used to confirm RPE necrosis in this model. Retinal flatmount revealed the appearance of PI-positive RPE cells as early as 24 h after NaIO3 injection (Figures 3e–h). At 48 h post injection, most RPE cells were PI-positive. The PI staining was observed in both the cytoplasm and nucleus of the RPE cells. A similar effect was observed at 72 h post injection. This DAPI/PI staining suggests RPE necrosis within 72 h of retro-orbital NaIO3 injection.
Next, we used a combination of TUNEL assay and active caspase-3 staining to examine the nature of RPE cell death in this model. TUNEL-positive photoreceptor cells (Figures 3i–l) were abundantly detected as early as 24 h post NaIO3 injection, and persisted at 72 h. TUNEL-positive RPE cells were detected at 24 h, and appeared to be more abundant at 48 h. TUNEL-positive RPE cells decreased at 72 h (Figures 3i–l, arrowheads). However, active caspase-3 staining was observed only in photoreceptor layer but not in RPE cells (Figures 3m–p, arrowheads).
Taken together, RPE cells in the retro-orbital NaIO3 model showed loss of ZO-1 staining, positive PI and TUNEL staining, and negative active caspase-3 staining. These features indicate that RPE cells die from necrosis but not apoptosis in vivo in response to oxidative injury induced by low dose NaIO3.
Sodium iodate induces RIPK3 aggregation and RPE necroptosis
RIPK3 aggregation and the formation of the necrosome is a critical step in necroptosis. Although we and others have successfully established RIPK3 aggregation as a necrotic hallmark in vitro, detection of RIPK3 aggregation in vivo has been challenging. To further confirm whether RPE cells undergo necroptosis in vivo, we have made transgenic mouse lines expressing human RIPK3 fused to a GFP protein under the control of the RPE-specific VMD2 promoter (Figure 4A). The expression of RIPK3-GFP in the mouse lines was verified by western blot (Figure 4B).

Characterization of the necroptosis in vivo by using pVMD-RIPK3-GFP transgenic mice and by visualizing HMGB1 release. (A) Schematics of the construct for transgenic mice. (B) Confirmation of transgene expression in three lines (L1-L3) by western blot. (C) RIPK3-GFP expression in the RPE layer of RIPK3-Tg mice was visualized by GFP staining (a), RIPK3-GFP aggregation as a result of 20 mg/kg NaIO3 was visible at 24 and 48 h (b and c) post administration. Very few GFP staining was observed at 72 h (d) (n=5 each). The scale bar is 10 μM. (D) Release of HMGB1 from the nucleus of RPE cells was visualized by HMGB1 staining. In normal RPE cells, HMGB1 is located in the nucleus (a), and 24 h after 20 mg/kg NaIO3 administration, HMGB1 was released into the cytoplasm (b, arrowheads). At 48 and 72 h, extranuclear HMGB1 can be observed in the RPE cell layer (c and d, arrowheads) (n=5 each). The scale bar is 25 μM. (E) HMGB1 release was measured in the vitreous humor using ELISA (n=3 each). ***P<0.001.
Immunostaining using anti-GFP antibody confirmed the expression of RIPK3-GFP specifically in the RPE cell layer of the transgenic mice (Figure 4C (a)). RIPK3 aggregation in the cytoplasm as shown by GFP staining was observed at 24 and 48 h after retro-orbital injection (Figure 4C (b and c)). RIPK3 aggregation was not observed at 72 h post injection, consistent with the notion that RIPK3 aggregation is an early event in necroptosis (Figure 4C (d)).
Necroptosis is also associated with the release of HMGB1, a member of the DAMP molecules, to the cytoplasm and outside of the cells. In the normal RPE cells, HMGB1 is localized in the nucleus (Figure 4D (a)). At 24 h post retro-orbital NaIO3 administration, extranuclear HMGB1 was detected in the RPE cell layer by immunostaining (Figure 4D (b-d)). Consistently, the level of HMGB1 was also significantly increased in the vitreous humor of the treated mice compared with the control by ELISA analyses (Figure 4E).
These experiments established that NaIO3 induces RIPK3 aggregation and HMGB1 release in mouse RPE cells in vivo, providing strong evidence of RPE necroptosis in this model.
Necrostatin-1 protects RPE from degeneration induced by NaIO3 in vivo
Our in vitro and in vivo data have established that RPE cells die from necroptosis after exposure to NaIO3. To further confirm RPE necroptosis in response to oxidative stress, we tested whether Nec-1, a potent and selective RIPK1 inhibitor, can inhibit RPE cell death in this model. Nec-1 (400 μM) was injected by retro-orbital injection at the time of NaIO3 administration. Mouse eyes were harvested 48 h post Nec-1/NaIO3 administration. Morphological analysis revealed that Nec-1 protected RPE cells from degeneration and partially maintained organization of the inner segments of the photoreceptors (Figures 5a–c). Nec-1 significantly decreased TUNEL positivity in RPE cells, while photoreceptors retained TUNEL positivity, suggesting that photoreceptor cell death could occur independently of RPE cell death in response to oxidative stress in this model (Figures 5d–f). Collectively, our data show that Nec-1 inhibits RPE cell death in vivo, which indicates that RPE cells die predominantly from necroptosis in response to NaIO3.

Inhibitor of necroptosis prevents RPE degeneration. (a–c) Hematoxylin and eosin staining of paraffin-embedded retinal cross sections at 48 h post 20 mg/kg NaIO3 or NaIO3/Nec-1 retro-orbital administration. (d–f) TUNEL analysis of RPE/photoreceptor cell death. The scale bar is 25 μM. n=6 each.
Discussion
The oxidizing agent NaIO3 has been known to induce selective toxicity in RPE cells. Here, we show that RPE cells die mainly from necroptosis in response to NaIO3 in vitro and in vivo. We found that NaIO3 induces hallmarks of necroptosis in RPE cells in vitro, such as RIPK3 aggregation and HMGB1 nuclear release. RIPK1 and RIPK3 inhibitors, but not pan-caspase or pyroptosis inhibitors, prevent NaIO3-induced RPE cell death. RPE cell necroptosis was confirmed in vivo by TUNEL assay, active caspase-3, and HMGB1 staining in NaIO3-induced RPE degeneration model. RIPK3 aggregation in response to NaIO3 was observed in RPE using novel RIPK3-GFP mouse lines. Furthermore, Nec-1 inhibits RPE cell death in response to NaIO3 in vivo. Our results indicate that RPE cells die mainly from necroptosis in response to oxidizing agents, which may have implications in the mechanism and therapeutics of dry AMD, especially GA.
RPE necroptosis in RPE cells in response to sodium iodate in vitro
Owing to the critical role of oxidative stress in AMD, numerous efforts have been focused on studying the mechanism of oxidative stress-induced RPE cell death as an approach to decipher the mechanism of AMD pathogenesis. Using either an H2O2 or tBHP model, researchers attributed oxidative stress-induced RPE cell death mostly to apoptosis. Our published data have shown that H2O2 and tBHP, induce oxidative stress and RPE necroptosis in vitro.17 Here, we extended our study to analyze RPE cell death in response to NaIO3, a chemical that induces reactive oxygen species production and is selectively toxic to RPE cells.23,27 We observed hallmarks of necroptosis in RPE cells in response to NaIO3 in vitro, including PI membrane permeability, RIPK3 activation, and HMGB1 release from the nucleus. These results are consistent with our results from H2O2 and tBHP treatments. Furthermore, we discovered that 200 μM Nec-1 or Nec-5, which inhibit RIPK1 directly or indirectly, respectively, protect NaIO3-induced RPE cell death. This observation is consistent with a previous report by Balmer et al.18 However, Nec-7, which targets other necrotic pathways independent of RIPK1, fails to rescue the cells. At the molecular level, activation of RIPK1 and RIPK3 signaling is a hallmark of necrosis.28–30 Upon induction of necrosis, RIPK3 forms a complex with RIPK1 that results in necrosome formation. NaIO3 induced distinct points of RIPK3 aggregation localized in cell periphery. Use of an RIPK3-specific inhibitor GSK’872 further confirmed the involvement of RIPK3 in NaIO3-mediated necroptosis. These observations suggest that NaIO3-mediated RPE cell death is dependent on RIPK1 and RIPK3.
RIPK3 aggregation and activation have been recently shown to activate the inflammasome under certain conditions,20,31 and inflammasome activation has been implicated in the AMD pathogenesis.32–35 Inflammasome activation could lead to the induction of pyroptosis, a type of cell death that depends on caspase-1 activation. Canonical inflammasome activation was not detected by ASC-GFP visualization, while a caspase-1-specific inhibitor Ac-YVAD failed to rescue NaIO3-induced RPE cell death, arguing against pyroptosis under these conditions. On the other hand, pan-caspase inhibitor z-VAD experiments argue against significant apoptosis under NaIO3 treatment. Taken together, our data support the induction of RIPK1/RIPK3-dependent necroptosis in RPE cells under oxidative stress.
Mechanism of RPE cell death response to sodium iodate and oxidative stress in vivo
There have been numerous histological studies on the in vivo effect of NaIO3 in different animal models.26,36–39 Damage to the retina after NaIO3 administration has been shown to be dose-, route-, and time-dependent. Morphological examination of the retina after 100 mg/kg NaIO3 administration has indicated RPE depigmentation, swelling, and vacuolization, suggesting RPE necrosis in this model.23,40 Evaluation of different doses of NaIO3 (15–70 mg/kg) has shown a progressive increase in RPE and photoreceptor damage with increasing dosage, and the dose below 10 mg/kg has been shown to have little effect on the retina.24,41–43 The common histopathological changes associated with NaIO3 administration include: discontinuity of the RPE layer, disrupted structure of the outer and inner photoreceptor segments, and macrophage infiltration. Destruction of the blood–retina barrier visualized as degradation of ZO-1 protein has been previously described as a result of NaIO3 administrations in rabbits.44 It was also reported that the RPE damage is most pronounced in the central region of the retina, with relatively little degeneration in the peripheral retina.43 However, detailed molecular studies on how RPE cells die in this model have not been performed. We adopted a retro-orbital injection of low-dose NaIO3 (20 mg/kg) to study RPE cell death in mice, with the intent to capture the early molecular events leading to RPE death.24–26
Our morphological analyses showed that RPE cells start to lose pigmentation at 24 h, and appear swollen, vacuolated, and start breaking off from the RPE layer at 48–72 h post NaIO3 administration. More severe RPE damage was observed in the central region of the retina compared with the peripheral regions. We also used in vivo PI staining, TUNEL assay, and active caspase-3 staining to distinguish between necrosis and apoptosis in RPE and photoreceptor cells. We found that mouse RPE cells become PI-positive and TUNEL-positive but cleaved caspase-3-negative, while photoreceptors became TUNEL- and cleaved caspase-3-positive. These observations suggest RPE necroptosis and photoreceptor apoptosis in response to NaIO3 in vivo. RPE necroptosis in this model was further confirmed by HMGB1 release from the nucleus and RIPK3 aggregation using our pVMD2-RIPK3-GFP indicator mice. Moreover, Nec-1 was able to rescue RPE but not photoreceptor cell death in vivo in this model. Taken together, our data provide compelling evidence that RPE cells die mainly from necroptosis in response to NaIO3 in vivo.
Our data that RPE cells die from necroptosis in vitro and in vivo is consistent with histopathological analyses in several other AMD animal models and human AMD samples. The SOD family proteins constitute a major component of the antioxidant system. Senescent Sod1−/− mice displayed RPE vacuolization, one of the common morphological hallmarks of necrotic cells.45 Similarly, RPE hypopigmentation, vacuolization, and atrophy were observed in a ribozyme AAV virus-generated SOD2 knockdown mouse model.46 NRF2 regulates expression of many antioxidant/detoxification genes.47 Nrf2−/− mice develop age-dependent RPE degeneration, spontaneous CNV, and subretinal inflammatory protein deposits.48 The RPE cells were highly vacuolated with membranous debris. Carboxyethylpyrrole is a unique oxidation fragment of docosahexaenoic acid found in AMD drusen and in plasma samples from AMD patients.49,50 Carboxyethylpyrrole-MSA-immunized mice were recently established as a model for studying geographic RPE atrophy.51 Features of RPE necrosis, including vacuolization, swelling, cell lysis, and nuclear pyknosis were observed.
Clinicopathological studies have defined the RPE alterations in GA.52–54 RPE rounding, sloughing, and entrapment of RPE-derived fragments within basal deposits have been observed in GA samples, supporting necrosis as a mechanism for RPE cell death in GA. Future studies should focus on confirming the molecular nature of RPE cell death in AMD animal models and human AMD samples. RPE necroptosis may have significant implications in AMD. As evidenced in this study, necroptosis inhibitors, such as Nec-1, could potentially be used to prevent RPE death in AMD and GA.
Materials and Methods
Generation of RIPK3-GFP transgenic mouse lines
Human RIPK3 cDNA was amplified from the pcDNA3.1 plasmid encoding HA-3xFlag-RIPK3-GFP using the following primers: 5′- AAAAAAGTCGACATGGATTATAAAGATGATGATG-3′ and 5′- AAAAAAGTCGACGAGCTCTAGCATTTAGGTGCA-3′. After digestion with SalI, the purified PCR product was cloned into pVMD2-rtTA vector to replace rtTA.55 Linearized vector was used for transgenic injection. Three lines with germline transmission were obtained. These mice are viable, with RPE-specific transgene expression confirmed by western blot and GFP staining. Western blot was performed as described elsewhere.56 The antibodies used were: primary anti-GFP (1 : 1000, Aves, Trigard, OR, USA) and secondary IRDye 680RD donkey anti-chicken (1 : 5000, LiCor, Lincoln, NE, USA).
Sodium iodate injections
For retro-orbtial NaIO3 injection, six -week-old C57BL/6J mice of both sexes or pVMD2-RIP3-GFP mice were anesthetized with ketamine/xylazine cocktail; in addition, a topical analgesic was used with 0.5% proparacaine solution in PBS. Sterile freshly prepared 1% NaIO3 solution was used for injection via the retro-orbital sinus at 20 mg/kg body weight.57 Control mice were injected with PBS.
Histology and immunohistochemistry
Ocular tissue was fixed in weak formalin Davidson’s solution, embedded in paraffin, and sectioned at 5 μm. Paraffin was removed with orange terpenes (Histoclear, National Diagnostics, Atlanta, GA, USA) and tissue rehydrated through graded ethanol prior to downstream applications. For conventional pathology, tissue sections were stained with Weigert’s Iron Hematoxylin and Eosin Y. For immunostaining, ocular tissue was subjected to heat-mediated antigen retrieval using epitope recovery buffer (Electron Microscopy Sciences, Hatfield, PA, USA), washed with PBS, and permeablized with 0.2% Triton X-100 in PBS. After blocking in 10% normal goat serum, primary antibody was applied and samples were incubated overnight (anti-HMGB1, 1 : 100, Cell Signaling, Danvers, MA, USA; anti-cleaved caspase-3, 1 : 200, Cell Signaling) or for 48 h (Anti-GFP, 1 : 500, Aves) at 4 °C. Alexa Fluor 594-conjugated rabbit anti-mouse (1 : 800) or Alexa Fluor 488 goat anti-chicken (1 : 800) secondary antibodies (Life Technologies, Carlsbad, CA, USA) was incubated with tissue sections for 1 h at room temperature. After washing with PBS, tissue sections were mounted with DAPI containing mounting medium (Vector, Burlingame, CA, USA) and analyzed under fluorescence microscope.
Semi-thin section preparation and methylene blue staining
Enucleated mouse eye globes were fixed in weak formalin Davidson’s Solution for 4 h at room temperature, followed by soaking in 70% ethanol for 1 h. After removing tissue from ethanol, the eye was gross cut in the sagittal plane (removing ~20% of the globe) with the lens not removed. The tissue was further dehydrated through 95% and absolute ethanol. Next, the tissue was soaked in acrylic resin (Unicryl, BB International, Cardiff, UK) for 1 h at room temperature with shaking. After 1 h, the resin was removed, fresh resin was added, and the tissue was allowed to shake overnight at room temperature. The next day, the tissue was transferred to an embedding mold and fresh acrylic resin was added. The tissue was baked in the embedding mold for 48 h at 60 °C. After baking, 1-μm sections were cut with a glass knife on an ultra-microtome (Reichert Ultracut S, Leica Microsystems, Wetzlar, Germany). Sections were floated on drops of water on a microscope slide and baked on a slide warmer (~55 °C) until water evaporated. For staining, 0.2% methylene blue diluted in 95% ethanol was added to the sections while the slides were still on the slide warmer. After 15–30 s, slides were rinsed with deionized water, flooded with orange terpene (Histoclear, National Diagnostics), tapped dry on a paper towel, and mounted with permanent mounting medium (Permount, Fisher Chemical, Waltham, MA, USA).
TUNEL assay
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was used to analyze cell death in vivo.58 Briefly, paraffin was removed from the ocular sections by orange terpene (Histoclear, National Diagnostics) and rehydrated through a graded ethanol series to double distilled water. Tissue sections were then permeabilized in freshly prepared 0.1% Triton X-100, 0.1% sodium citrate solution. Terminal deoxynucleotidyl transferase solution was prepared according to manufacturer’s directions (Roche Applied Science, Mannheim, Germany) and incubated with the tissue sections for 60 min at 37 °C. Samples were then washed with PBS, mounted with DAPI containing mounting solution (Vector), and analyzed under the fluorescent microscope (Nikon, Tokyo, Japan).
Sclera-RPE flatmount ZO-1 and PI staining
For flatmount ZO-1 staining, enucleated mouse eye globes were washed in PBS. The anterior chamber, lens, and neuroretina were removed and the eyecup was fixed in 100% methanol for 30 min at room temperature. Next, the eyecup was incubated in blocking buffer containing 0.5% Triton and 5% horse serum (Gibco, Waltham, MA, USA) in PBS for 1 h at room temperature. Primary antibody against zonula occludens (ZO)-1 (1 : 100, Life Technologies) was incubated with the eyecup at 4 °C overnight. Alexa Fluor 594-conjugated goat anti-rabbit (1 : 800) was prepared in PBS and incubated with cells for 1 h at room temperature. After wash with 1× PBS, the eyecup was flat-mounted with DAPI containing mounting medium for fluorescence (Vector) and analyzed under a fluorescence microscope (Nikon). Flatmount PI staining was performed similarly as described elsewhere.59 PI (0.5 μg, Life Technologies) was injected through retro-orbital injection at 15 min before killing the mice. The eye globes were enucleated, anterior part was removed, and retinas were flat-mounted for fluorescence microscopy.
Cell culture, transfection, treatments, and MTT assay
Human RPE cells (ARPE-19, CLR-2302, ATCC, Manassas, VA, USA) were cultured in DME/F-12 medium (HyClone, Logan, UT, USA) supplemented with 10% FBS (HyClone) and 1× Penicillin-Streptomycin solution (HyClone) at 37 °C in 5% CO2. Cells were treated with: 50 μM z-VAD (Sigma-Aldrich, St Louis, MO, USA), 200 μM necrostatin-1, -5, -7 (Enzo Life Sciences, Farmingdale, NY, USA), 50 μM caspase-1 inhibitor Ac-YVAD (Sigma-Aldrich), or 3 μM GSK’872 (Millipore, Billerica, MA, USA). Cells were treated with 10 mM of sodium iodate (Sigma-Aldrich) in PBS (Gibco).
Cell transfection was performed using Lipofectamine LTX (Life Technologies, Farmingdale, NY, USA). Briefly, 1 μg of HMGB1-YFP, ANT1-RFP, RIPK3-GFP, or PYCARD-GFP plasmid DNA was mixed with 5 μl of Lipofectamine LTX. The complex was added to the ARPE-19 cell cultured in 4-chamber glass slide 20 min later. Expression of the recombinant proteins was visualized upon NaIO3 treatment after 24 h under a fluorescent microscope.
To assess cell viability, MTT assay was performed as described previously.60 In short, ARPE-19 cells were incubated with 1 mg/ml of MTT reagent (Sigma-Aldrich) for 2 h in standard cell culture conditions. Developed MTT crystals were dissolved in DMSO (Sigma-Aldrich) and analyzed at the 96-well plate reader by measuring absorbance at 540 nm.
Mouse vitreous humor collection and ELISA analysis
Vitreous humor collection was performed as described previously.61 In brief, a linear incision was made in the cornea and the anterior chamber fluid was removed. Next, pressure was applied at the external surface of the sclera, and the lens was pushed forward through the corneal incision. The vitreous gel was placed in the centrifugation tube and dissolved in PBS (Gibco). Level of HMGB1 in mouse vitreous humor was measured by enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s manual (Elabscience, WuHan, China). Briefly, the samples were added to the plate pre-coated with anti-HMGB1 antibody and incubated for 90 min at 37 °C. After removing the samples, biotinylated detection antibody was added. After incubation for 1 h at 37 °C and PBS wash, HRP conjugate was added and the samples were incubated for 30 min at 37 °C. After washing with PBS, substrate reagent was added to the plate for 15 min at 37 °C, and the reaction was stopped by the stop solution. The plate was analyzed on a micro-plate reader at 450 nm (Molecurar Devices, Sunnyvale, CA, USA).
Statistics
Each experiment was repeated at least three times. Student’s t-tests were used to determine statistical significance between groups. P-values of less than 0.05 were considered to be statistically significant.
Abbreviations
- AMD:
-
age-related macular degeneration
- ANT:
-
adenine nucleotide translocator
- ASC:
-
apoptosis-associated speck-like protein containing a carboxy-terminal CARD
- ATP:
-
adenosine triphosphate
- ELISA:
-
enzyme-linked immunosorbent assay
- GA:
-
geographic atrophy
- GFP:
-
green fluorescent protein
- H2O2:
-
hydrogen peroxide
- HMGB1:
-
high mobility group box 1
- NRF2:
-
nuclear factor (erythroid-derived 2)-like 2
- PI:
-
propidium iodide
- RFP:
-
red fluorescent protein
- RIPK:
-
receptor interacting protein kinase
- RPE:
-
retinal pigment epithelium
- SOD1:
-
superoxide dismutase 1
- tBHP:
-
tert-butyl hydroperoxide
- TUNEL:
-
terminal deoxynucleotidyl transferase dUTP nick end labeling
- YFP:
-
yellow fluorescent protein
- ZO-1:
-
Zona occludens 1.
References
Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 2004; 122: 564–572.
Ambati J, Fowler BJ . Mechanisms of age-related macular degeneration. Neuron 2012; 75: 26–39.
Cai X, McGinnis JF . Oxidative stress: the achilles’ heel of neurodegenerative diseases of the retina. Front Biosci 2012; 17: 1976–1995.
Age-Related Eye Disease Study 2 Research G. Lutein+zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 2013; 309: 2005–2015.
Age-Related Eye Disease Study Research G. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 2001; 119: 1417–1436.
Wang J, Lacovelli J, Spencer C, Saint-Geniez M . New insights on sodium iodate-induced retianl degeneration. FASEB J 2013; 27: Suppl 873.15.
Zulliger R Cellular and molecular changes in animal models of retinal degeneration [PhD]: University of Bern 2010.
Korte GE, Reppucci V, Henkind P . RPE destruction causes choriocapillary atrophy. Invest Ophthalmol Vis Sci 1984; 25: 1135–1145.
Noell WK . Experimentally induced toxic effects on structure and function of visual cells and pigment epithelium. Am J Ophthalmol 1953; 36: 103–116.
Baich A, Ziegler M . The effect of sodium iodate and melanin on the formation of glyoxylate. Pigment Cell Res 1992; 5: 394–395.
Konda BR, Pararajasegaram G, Wu GS, Stanforth D, Rao NA . Role of retinal pigment epithelium in the development of experimental autoimmune uveitis. Invest Ophthalmol Vis Sci 1994; 35: 40–47.
Ashburn Jr FS, Pilkerton AR, Rao NA, Marak GE . The effects of iodate and iodoacetate on the retinal adhesion. Invest Ophthalmol Vis Sci 1980; 19: 1427–1432.
Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 2007; 9: 49–89.
Christofferson DE, Yuan J . Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 2010; 22: 263–268.
Zitvogel L, Kepp O, Kroemer G . Decoding cell death signals in inflammation and immunity. Cell 2010; 140: 798–804.
Hanus J, Anderson C, Wang S . RPE necroptosis in response to oxidative stress and in AMD. Ageing Res Rev 2015; 24 (Pt B): 286–298.
Hanus J, Zhang H, Wang Z, Liu Q, Zhou Q, Wang S . Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis 2013; 4: e965.
Balmer J, Zulliger R, Roberti S, Enzmann V . Retinal cell death caused by sodium iodate involves multiple caspase-dependent and caspase-independent cell-death pathways. Int J Mol Sci 2015; 16: 15086–15103.
Qin S, Lu Y, Rodrigues GA . Resveratrol protects RPE cells from sodium iodate by modulating PPARalpha and PPARdelta. Exp Eye Res 2014; 118: 100–108.
Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 2015; 6: 6282.
Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 2007; 14: 1590–1604.
Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012; 149: 847–859.
Zhou P, Kannan R, Spee C, Sreekumar PG, Dou G, Hinton DR . Protection of retina by alphaB crystallin in sodium iodate induced retinal degeneration. PloS One 2014; 9: e98275.
Wang J, Iacovelli J, Spencer C, Saint-Geniez M . Direct effect of sodium iodate on neurosensory retina. Invest Ophthalmol Vis Sci 2014; 55: 1941–1953.
Xia H, Krebs MP, Kaushal S, Scott EW . Enhanced retinal pigment epithelium regeneration after injury in MRL/MpJ mice. Exp Eye Res 2011; 93: 862–872.
Machalinska A, Lubinski W, Klos P, Kawa M, Baumert B, Penkala K et al. Sodium iodate selectively injuries the posterior pole of the retina in a dose-dependent manner: morphological and electrophysiological study. Neurochem Res 2010; 35: 1819–1827.
Sorsby A . Experimental pigmentary degeneration of the retina by sodium iodate. Br J Ophthalmol 1941; 25: 58–62.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–336.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012; 148: 213–227.
Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D . Caspase-8 Blocks Kinase RIPK3-Mediated Activation of the NLRP3 Inflammasome. Immunity 2013; 38: 27–40.
Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med 2012; 18: 791–798.
Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K . Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells--implications for age-related macular degeneration (AMD). Immunol Lett 2012; 147: 29–33.
Marneros AG . NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Cell Rep 2013; 4: 945–958.
Wang Y, Hanus JW, Abu-Asab MS, Shen D, Ogilvy A, Ou J et al. NLRP3 upregulation in retinal pigment epithelium in age-related macular degeneration. Int J Mol Sci 2016; 17: pii: E73.
Deberardinis E, Vecchione L, Tieri O . Studies of the mechanism of the action of the sodium iodate on the rabbit retina. Acta Ophthalmol (Copenh) 1964; 42: 713–718.
Tamaki S . [The influence of sodium iodate on ERG of rats]. Nihon Ganka Kiyo 1968; 19: 566–571.
Nilsson SE, Knave B, Persson HE . Changes in ultrastructure and function of the sheep pigment epithelium and retina induced by sodium iodate. II. Early effects. Acta Ophthalmol (Copenh) 1977; 55: 1007–1026.
Murray MM . The effects of administration of sodium iodate to man and animals. Bull World Health Organ 1953; 9: 211–216.
Kiuchi K, Yoshizawa K, Shikata N, Moriguchi K, Tsubura A . Morphologic characteristics of retinal degeneration induced by sodium iodate in mice. Current Eye Res 2002; 25: 373–379.
Franco LM, Zulliger R, Wolf-Schnurrbusch UE, Katagiri Y, Kaplan HJ, Wolf S et al. Decreased visual function after patchy loss of retinal pigment epithelium induced by low-dose sodium iodate. Invest Ophthalmol Vis Sci 2009; 50: 4004–4010.
Machalinska A, Lejkowska R, Duchnik M, Kawa M, Roginska D, Wiszniewska B et al. Dose-dependent retinal changes following sodium iodate administration: application of spectral-domain optical coherence tomography for monitoring of retinal injury and endogenous regeneration. Curr Eye Res 2014; 39: 1033–1041.
Machalinska A, Lubinski W, Klos P, Kawa M, Baumert B, Penkala K et al. Sodium iodate selectively injuries the posterior pole of the retina in a dose-dependent manner: morphological and electrophysiological study. Neurochem Res 2010; 35: 1819–1827.
Ringvold A, Olsen EG, Flage T . Transient breakdown of the retinal pigment epithelium diffusion barrier after sodium iodate: a fluorescein angiographic and morphological study in the rabbit. Exp Eye Res 1981; 33: 361–369.
Hashizume K, Hirasawa M, Imamura Y, Noda S, Shimizu T, Shinoda K et al. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am J Pathol 2008; 172: 1325–1331.
Justilien V, Pang JJ, Renganathan K, Zhan X, Crabb JW, Kim SR et al. SOD2 knockdown mouse model of early AMD. Invest Ophthalmol Vis Sci 2007; 48: 4407–4420.
Zenkov NK, Menshchikova EB, Tkachev VO . Keap1/Nrf2/ARE redox-sensitive signaling system as a pharmacological target. Biochemistry 2013; 78: 19–36.
Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE et al. Age-related retinopathy in NRF2-deficient mice. PLoS One 2011; 6: e19456.
Gu X, Meer SG, Miyagi M, Rayborn ME, Hollyfield JG, Crabb JW et al. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem 2003; 278: 42027–42035.
Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA 2002; 99: 14682–14687.
Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med 2008; 14: 194–198.
Sarks SH . Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol 1976; 60: 324–341.
Sarks JP, Sarks SH, Killingsworth MC . Evolution of geographic atrophy of the retinal pigment epithelium. Eye (London, England) 1988; 2 (Pt 5): 552–577.
Rudolf M, Vogt SD, Curcio CA, Huisingh C, McGwin Jr G, Wagner A et al. Histologic basis of variations in retinal pigment epithelium autofluorescence in eyes with geographic atrophy. Ophthalmology 2013; 120: 821–828.
Le YZ, Zheng W, Rao PC, Zheng L, Anderson RE, Esumi N et al. Inducible expression of cre recombinase in the retinal pigmented epithelium. Invest Ophthalmol Vis Sci 2008; 49: 1248–1253.
Hanus J, Zhang H, Chen DH, Zhou Q, Jin P, Liu Q et al. Gossypol acetic acid prevents oxidative stress-induced retinal pigment epithelial necrosis by regulating the FoxO3/Sestrin2 pathway. Mol Cell Biol 2015; 35: 1952–1963.
Yardeni T, Eckhaus M, Morris HD, Huizing M, Hoogstraten-Miller S . Retro-orbital injections in mice. Lab Anim (NY) 2011; 40: 155–160.
Li X, Zhou Q, Hanus J, Anderson C, Zhang H, Dellinger M et al. Inhibition of multiple pathogenic pathways by histone deacetylase inhibitor SAHA in a corneal alkali-burn injury model. Mol Pharm 2013; 10: 307–318.
Unal Cevik I, Dalkara T . Intravenously administered propidium iodide labels necrotic cells in the intact mouse brain after injury. Cell Death Differ 2003; 10: 928–929.
Hanus J, Kolkin A, Chimienti J, Botsay S, Wang S . 4-Acetoxyphenol prevents RPE oxidative stress-induced necrosis by functioning as an NRF2 stabilizer. Invest Ophthalmol Vis Sci 2015; 56: 5048–5059.
Skeie JM, Tsang SH, Mahajan VB . Evisceration of mouse vitreous and retina for proteomic analyses. J Vis Exp 2011; (50):pii: 2795.
Acknowledgements
We would like to thank: Dr. Qinghua Liu from University of Texas Southwestern Medical Center for providing ASC-GFP plasmid; Dr. Zhigao Wang from University of Texas Southwestern Medical Center for providing HA-3xFlag-RIPK3-GFP, HMGB1-YFP, and ANT1-RFP plasmids; Dr. Yunzheng Le from Oklahoma Health Science Center for providing pVMD2-rtTA plasmid; Dr. Yiping Chen from Tulane University for helpful discussions; Dr. Frank Jones from Tulane University for sharing equipment; and Julia Chimienti for help with the experiments. This research was supported by a startup fund from Tulane University, NIH Grant EY021862, a career development award from the Research to Prevent Blindness foundation, and a Bright Focus Foundation Award in Age-related Macular Degeneration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Rufini
Supplementary Information accompanies the paper on the Cell Death and Discovery website (http://www.nature.com/cddiscovery)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hanus, J., Anderson, C., Sarraf, D. et al. Retinal pigment epithelial cell necroptosis in response to sodium iodate. Cell Death Discovery 2, 16054 (2016). https://doi.org/10.1038/cddiscovery.2016.54
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/cddiscovery.2016.54
This article is cited by
-
Involvement of FSP1-CoQ10-NADH and GSH-GPx-4 pathways in retinal pigment epithelium ferroptosis
Cell Death & Disease (2022)
-
Morphologic and electrophysiologic findings of retinal degeneration after intravitreal sodium iodate injection following vitrectomy in canines
Scientific Reports (2020)
-
Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics
Nature Communications (2020)
-
Inhibition of thyroid hormone signaling protects retinal pigment epithelium and photoreceptors from cell death in a mouse model of age-related macular degeneration
Cell Death & Disease (2020)
-
D609 protects retinal pigmented epithelium as a potential therapy for age-related macular degeneration
Signal Transduction and Targeted Therapy (2020)

{kind=link}