
Abstract
The mechanisms of how signaling pathways are coordinated and integrated for the maintenance of the self-renewal of human embryonic stem cells (hESCs) and the acquisition of pluripotency in reprogramming are still only partly understood. CDK1 is a key regulator of mitosis. Recently, CDK1 has been shown to be involved in regulating self-renewal of stem cells, even though the mechanistic role of how CDK1 regulates pluripotency is unknown. In this report, we aim to understand how CDK1 can control pluripotency by reducing CDK1 activity to a level that has no effect on cell cycle progression. We demonstrated that high levels of CDK1 is associated with the pluripotency stage of hESCs; and decreased CDK1 activity to a level without perturbing the cell cycle is sufficient to induce differentiation. CDK1 specifically targets the phosphorylation of PDK1 and consequently the activity of PI3K/Akt and its effectors ERK and GSK3β. Evidence of the reversion of inactive CDK1-mediated differentiation by the inhibition of Akt signaling effectors suggests that the CDK1-PDK1-PI3K/Akt kinase cascade is a functional signaling pathway for the pluripotency of hESCs. Moreover, cyclin B1-CDK1 complexes promote somatic reprogramming efficiency, probably by regulating the maturation of induced pluripotent stem cells (iPSCs), as cyclin B1 stimulates a higher cellular level of LIN28A, suggesting that monitoring iPSC factors could be a new path for the enhancement of reprogramming efficiency. Together, we demonstrate an essential role for the CDK1-PDK1-PI3K/Akt kinase signaling pathway in the regulation of self-renewal, differentiation, and somatic reprogramming, which provides a novel kinase cascade mechanism for pluripotency control and acquisition.
Similar content being viewed by others


Main
CDK1 is one of the most pleiotropic cell cycle regulators; it not only primarily interacts with cyclin B to drive the G2-M transition but also binds to other interphase cyclins (cyclin D1, E, and A) to regulate G1 progression and G1-S transition.1 Cdk1 alone is sufficient to drive mammalian cell cycle progression in knockout mice lacking other Cdks, indicating that Cdk1 can compensate for other interphase Cdks during embryonic development.2 However, liver-specific deletion of Cdk1 is well tolerated and does not impair liver regeneration.3 Cdk1 is also essential for meiosis in mouse oocytes,4 and the suppression of Cdk1 leads to the differentiation of mouse trophoblast stem cells into giant cells.5 Recently, it has been demonstrated that Cdk1/CDK1 is required for self-renewal in both mESCs and human embryonic stem cells (hESCs),6, 7 which might be related to its interaction with Oct4.8, 9 CDK1/CDK2 potentially regulates a large number of substrates (at least 1220) during hESC differentiation.10 All of these studies point to the association of CDK1 with pluripotency, although there has been no study demonstrating the mechanistic role of how CDK1 regulates pluripotency.
Self-renewal and pluripotency of ESCs are maintained by auto-regulatory networks involving the core transcriptional factors NANOG, OCT4, and SOX2, as well as chromatin remodeling complexes and epigenetic modifiers.11 Moreover, the activation of pluripotency genes and the suppression of lineage-determined genes require the integration of multiple internal and external signaling pathways of which the developmental differences between mESCs and hESCs could affect their differential responses to signaling regulation.12 In hESCs, NODAL/ACTIVIN induces SMAD2/3 signaling and the key target gene NANOG for self-renewal. NODAL/ACTIVIN together with hyperactive PI3K/Akt signaling, which is stimulated by the growth factors bFGF and IGFs, suppresses ERK activity and dephosphorylation of GSK3β, these pathways contribute to the maintenance of pluripotency.13, 14, 15, 16 In contrast to hESCs, the primary function of PI3K/Akt in naive mESCs is to suppress Gsk3β activity through Gsk3β hyperphosphorylation, which allows Nanog and c-Myc to maintain pluripotency.17 So far, only a few cell signaling pathways have been shown to be important for hESC pluripotency, and a possible link between the cell signaling pathways and basic cellular properties, such as cell cycle and cell cycle regulators, has not been well addressed. Here, we investigate the role of CDK1 in the biology of hESCs. In addition to being a critical cell cycle regulator, our results identify the novel CDK1-PDK1-PI3K/Akt kinase cascade as an important signaling pathway for the control and acquisition of pluripotency.
Results
High levels of CDK1 is associated with the pluripotency stage of hESCs
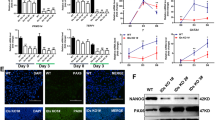
Cdk1 is indispensable and cannot be compensated by interphase Cdks during early embryonic development,2, 3 indicating a potential in controlling pluripotency in addition to its function as a cell cycle regulator. However, the existence of a direct association between CDK1 and pluripotency state has not been addressed. To understand this association, we found that hESCs contained a high level of CDK1. Upon embryoid body (EB)- and retinoic acid-mediated hESC differentiation (the enhanced expression of several lineage markers confirmed differentiation; Supplementary Figures S1a and b), downregulation of pluripotency factors NANOG, OCT4, and SOX2 was accompanied by a decrease of CDK1 at both the mRNA and protein levels (Figures 1a–d and Supplementary Figure S1c). The expression of other cell cycle regulators such as CDK2 remained unchanged (Figure 1b). A correlation between the downregulation of pluripotency markers and CDK1 was also observed in human NCCIT during EB- and retinoic acid-mediated differentiation (Supplementary Figure S1d). In differentiated EBs, CDK1 was decreased to a level that was associated with differentiation but retained the ability to maintain proliferation (Supplementary Figure S1b). Thus, CDK1 is not only essential for early embryonic development as a cell cycle driver, but is also associated with the undifferentiated state of hESCs.

High CDK1 expression is correlated with hESC pluripotent state. (a and b) During EB-mediated differentiation of hESCs, CDK1 expression decreases in parallel with pluripotency genes NANOG, OCT4, and SOX2 as measured by qRT-PCR (a) and immunoblot (b). (c) qRT-PCR and immunoblot. (d) Measurement of NANOG, OCT4, SOX2, and CDK1 expression in FBS or retinoic acid-mediated hESC differentiation. qRT-PCR data are represented as the mean±S.D.; n=2, each in duplicate. (e) Transient knockdown of NANOG or OCT4 by lentiviral shRNA in hESCs followed by immunoblotting for NANOG, OCT4, and CDK1. (f) Downregulation of CDK1 is associated with a decrease in NANOG and OCT4 during retinoic acid-mediated differentiation. The CDK1 level presented by the histogram was gated from NANOG+high and NANOG− population and OCT4+high and OCT4− population, respectively. (g) Decreased NANOG and OCT4 levels might also be associated with the downregulation of CDK1 in retinoic acid-mediated differentiation. Histogram levels of NANOG and OCT4 were gated from CDK1+high and CDK1+low populations
The decreased expression of CDK1 was detected after lentiviral shRNA-mediated suppression of pluripotency factors NANOG and OCT4 (Figure 1e). Thus, when hESCs lost their pluripotency, the corresponding downregulation of CDK1 suggests a true association between CDK1 and the pluripotency state. During hESC differentiation, the expression level of CDK1 was coupled with NANOG and OCT4, with a higher level of CDK1 found only in NANOG+high and OCT4+high populations (Figure 1f). Conversely, CDK1 expression also associated with NANOG and OCT4 expression, as lower expression of NANOG and OCT4 was found in cells with downregulated CDK1 (Figure 1g). Taken together, we demonstrated that the expression level of CDK1 is associated with pluripotency states.
Downregulation of CDK1 impairs pluripotency and promotes differentiation
Further to understand CDK1’s role in hESCs, we decreased the activity of CDK1 either by lentiviral shCDK1 transduction for 4 days or by treatment with the CDK1 inhibitor, RO3306 (8 μM) for 2 days. The mesoendoderm marker transcripts BRACHYURY (BRA), EOMES, GOOSECOID (GSC), and MIXL1 significantly increased (Figure 2a, left panel). In contrast, the pluripotency gene transcripts NANOG, OCT4, and SOX2 (Figure 2a, right panel), as well as the protein levels of NANOG, OCT4, SOX2, and SSEA4 (Figure 2b) significantly decreased. Analysis of hESC colonies with positive alkaline phosphatase activity demonstrated that self-renewal capacity was impaired after shCDK1 or RO3306 treatment (Figure 2c and Supplementary Figure S2a). To further test the effects of CDK1 inhibition on teratocarcinoma formation in vivo in NCCIT cells, we found that JNJ-770621 (a CDK1 inhibitor, 0.5 μM) reduced teratocarcinoma formation from 86 to 31% in SCID mice (Supplementary Figure S2b). These results suggest that downregulation of CDK1 impaired self-renewal and triggered differentiation in hESCs and NCCIT.

Downregulation of CDK1 promotes differentiation. (a) Transient knockdown of CDK1 by lentiviral shRNA or inactivation of CDK1 by RO3306 (8 μM) resulted in enhanced mesoendoderm markers (left panel) and decreased pluripotency transcripts (right panel). qRT-PCR data are represented as the mean±S.D., n=3, each in duplicate. A statistical comparison was made between shCtrl and shCDK1 or Ctrl and RO3306 by paired Student’s t-test (*P<0.05; **P<0.01; ***P<0.0001). (b) Flow cytometry data show a reduced expression of the pluripotency proteins NANOG, OCT4, SOX2, and SSEA4 at 2 days after RO3306 (8 μM) treatment. (c) Statistical comparison of alkaline phosphatase(+) colonies in shCDK1 knockdown or RO3306-treated hESCs. The data are represented as the mean±S.D. from three independent experiments. (d) BrdU-labeled cell cycle analysis of hESCs that were transiently transduced with lentiviral shCtrl or shCDK1 for 3 days. (e) Flow cytometry analysis of the pluripotency marker TRA-1-60 in hESCs that were treated with or without 8 μM of RO3306. (f) TRA-1-60 level gating from the live and early apoptotic populations in the RO3306-treated group from (e). (g) Flow cytometry analysis of NANOG expression in G1, S, and G2/M phases in hESCs that were treated with or without RO3306
Downregulation of CDK1 impairing pluripotency is not cell cycle and apoptosis regulated
We next investigated whether the impaired self-renewal of hESCs caused by CDK1 inactivation was through the traditional role of CDK1 in regulating the cell cycle and apoptosis. Transient knockdown of CDK1 by shRNA for 3–4 days did not result in changes in cell cycle distribution compared with shControl-tranducted hESCs (Figure 2d and Supplementary Figure S3a). A different sequence design was also applied to shCDK1 to exclude off-target effects, which also did not cause cell cycle arrest (Supplementary Figures S3b and c). Similarly, there was no significant cell cycle arrest in hESCs after incubation with RO3306 (8 μM) for 16–28 h, except with a minor reduction of S phase populations (8%) (Supplementary Figures S3a, d and e). Thus, the reduction of CDK1 to a level that does not cause a cell cycle arrest was able to promote differentiation of hESCs. However, the current study cannot exclude the possibility that the long-term downregulation of CDK1 might affect differentiation and proliferation via cell arrest.
Interrelation of differentiation, proliferation, and apoptosis in CDK1-inactivated hESCs
We investigated the correlation between differentiation and apoptosis by measuring a pluripotency cellular surface marker (TRA-1-60) in live, early apoptotic, and dead populations. The overall survival was comparable between lentiviral shControl- and shCDK1-transfected hESCs; with or without treatment with RO3306 (2 μM) (Supplementary Figure S4a). Similar results were obtained using NCCIT cells (Supplementary Figure S4b). RO3306 (8 μM) treatment for 26 h, the time course with no significant cell cycle arrests, and a reduced expression of TRA-1-60 was observed (Figure 2e). By monitoring the TRA-1-60 expression in different cell populations, we found that the ratio of hESCs with negative or low TRA-1-60 was significantly higher in the early apoptotic population than in the live population (Figure 2f and Supplementary Figure S4c); although it was concurrent with the slightly enhanced apoptotic population (compare Supplementary Figure S4a with d). We postulated that inactivation of CDK1 resulted in the loss of pluripotency markers at early time points when cell cycle perturbation was not a major factor. Inactivation of CDK1-related differentiation exerted further impact on proliferation and apoptosis of hESCs at later time points. However, cell cycle interruption, particularly the reduction of S and induction of G2 arrest, might occur under longer CDK1 perturbation. To monitor cell cycle phase-dependent differentiation, NANOG expression in G1, S, and G2/M phases was determined. The inactivation of CDK1 led to a shift of NANOG expression (Supplementary Figure S4e). NANOG expression was repressed in the G1 phase in comparison with that in the S and G2 phases. This was not because inactivation of CDK1 resulted in G1 arrest. Moreover, there was no significant difference in NANOG reduction between the S and G2 phases (Figure 2g). Together, cell cycle perturbation was not necessary grounds for ESC differentiation; but differentiation would further affect cellular proliferation and apoptosis.
Impact of CDK1 downregulation on PDK1 phosphorylation
As CDK1 was associated with the pluripotency state in an induced differentiation background, this led us to hypothesize a direct role of CDK1 in monitoring self-renewal. Predicted kinase cascades in early hESCs differentiation have linked CDK1/CDK2 to the regulation of protein kinase N,10 which is a substrate of CDK1 in vitro (www.phosphosite.org). Protein kinase N belongs to the AGC kinase family and is phosphorylated by 3′-phosphoinositide-dependent kinase-1 (PDK1), a master kinase for the activation of other AGC kinases, including Akt.18 To explore the potential link between CDK1 and PDK1, we found that in CDK1 knockdown hESCs, phosphorylation of PDK1 (Ser241), which is autophosphorylated and necessary for PDK1 activity,19 was notably reduced or diminished compared with that in shRNA control-transfected cells (Figure 3a). Upon the inactivation of CDK1 with RO3306 and JNJ-7706621 (1 μM), which are specific for CDK1 and had no direct effect on PDK1,20, 21 the phosphorylation of PDK1 was also significantly reduced (Figure 3b). These results suggest that the suppression of PDK1 phosphorylation could be the primary effect of CDK1 downregulation. In a bioinformatic analysis of human PDK1, a putative CDK1 phosphorylation site was identified at Thr354 (TPPK), which matches the CDK consensus phosphorylation motif (S/T-P-X-K/R, where X is any amino acid) and is evolutionarily conserved among human, mouse, and Xenopus PDK1 (Figure 3c). To determine the association between CDK1 and PDK1, co-immunoprecipitation analyses revealed that CDK1 interacted with PDK1 in vitro (Figure 3d). To further understand whether Thr354 could be phosphorylated by CDK1, a CDK1 kinase assay was performed using PDK1 Thr354 wild-type peptide and Thr354A mutant peptide as substrates. Fluorescence signals indicating ADP generated by CDK1 kinase activity was significantly higher when using Thr354 as a substrate than using Thr354A (P=0.03 in H7 and P=0.004 in NCCIT cells) (Figure 3e). Together, these results suggest that PDK1 is a potential novel functional substrate of CDK1 and CDK1 inactivation results in the suppression of PDK1 activity.

Inactivation of CDK1 specifically modifies the phosphorylation of PDK1 and Akt. (a) The phosphorylation of PDK1 (Ser241) after shCDK1 transduction in hESCs H1 and H7 was analyzed by immunoblotting. (b) Inactivating CDK1 by RO3306 and JNJ-7706621 results in decreased PDK1 phosphorylation in hESCs and NCCIT cells. (c) A CDK1 consensus phosphorylation site S/T-P-X-K/R in PDK1 at Thr354 (TPPK) is evolutionarily conserved among human, mouse, and Xenopus. S/T, serine/threonine; P, proline; X, any amino acid; K/R, lysine/arginine. (d) NCCIT cell lysates were immunoprecipitated with CDK1 antibody; the bound proteins were analyzed using PDK1 antibody. The loading was tested by immunoblotting on total PDK1 and CDK1 from equal amount lysates used for IP. (e) CDK1 antibody-immunoprecipitates were subjected to kinase assay using ADPsensor universal kinase activity assay kit. PDK1 T354 or T354A peptides were used as substrates. Incubating the peptide alone without CDK1 immunoprecipitates was used as background controls. Relative fluorescence unit (RFU) signals (after subtracting background) were calculated as ΔRFU (RFU2-RFU1) from different reaction time. *P<0.05, **P<0.01. (f) Transient knockdown of CDK1 in three lines of hESCs suppresses the phosphorylation of Akt at Thr308 and Ser473. (g) The phospho-(Ser/Thr) PDK1 docking motif was immunoprecipitated in shCDK1- or shControl-transduced hESCs. The immunoprecipitates were then analyzed using the antibody to phospho-Akt (Ser473). (h) The same analysis as in (f) using NCCIT cells that were treated with RO3306 (5 μM) for 24 h
Downregulation of CDK1 suppresses the PDK1-PI3K/Akt pathway
Phosphorylation of Akt at two critical phosphorylation sites (Thr308 and Ser473) is required for Akt activation. While Thr308 is phosphorylated by PDK1, Ser473 is activated directly by mTORC218 and indirectly by PDK1.22, 23 We observed that knockdown of CDK1 in three lines of hESCs was accompanied by a decrease of phosphorylation of Akt at Thr308 and Ser473 (Figure 3f), presumably due to the CDK1 inactivation-mediated suppression of PDK1 (Figure 3a). We noticed a limited reduction of Akt phosphorylation in CDK1-inactivated hESCs, probably because hESC culture medium contains high levels of Akt activators.13
Although the CDK1 consensus phosphorylation site on PDK1 (Thr354) is a phosphorylated site, it might not be related to the inhibition of PDK1 Ser214 phosphorylation after CDK1 inactivation. Thus, we performed co-immunoprecipitation using an antibody against the phospho-Ser/Thr docking motif of the PDK1, which covers all serine or threonine phosphorylating motifs, including Thr354, to co-precipitate the active form of Akt. Considering in addition to Ser214, other phosphorylated serine or threonine of PDK1 may contribute to endogenous levels of phospho-Akt. In CDK1 knockdown hESCs (Figure 3g) and CDK1-inactivated (by RO3306) NCCIT cells (Figure 3h), the phospho-Ser/Thr docking motif of PDK1 was inhibited and consequently precipitated less active form of Akt including pAkt on Ser473 than control cells. These data demonstrate not only a critical link between CDK1 and PDK1 but also a potential kinase cascade mode of action of the CDK1-PDK1-Akt signaling pathway. The fact that the CDK1-PDK1 pathway was able to regulate Akt signaling in hESCs suggests that in addition to growth factors,13 kinase signaling is also important and required for controlling PI3K/Akt signaling pathways for pluripotency.
Functional CDK1-PDK1-Akt kinase pathway is required for pluripotency
Following CDK1 knockdown, the suppressed PDK1-mediated decrease of Akt phosphorylation was observed even in the mTeSR1 medium, which contains growth factors for Akt activation. Furthermore, a low dose of CDK1 inhibitor (RO3306) did not directly inhibit Akt phosphorylation (Supplementary Figure S5a). Therefore, it would be critical to understand if CDK1 inactivation affected the effectors of the PI3K/Akt signaling pathway. We investigated the phosphorylation status of ERK1/2, GSK3β, and SMAD2/3. In response to reduced Akt phosphorylation after inactivation of CDK1, the phosphorylation of ERK1/2 (T202, Y204) and GSK3β (Ser9) was enhanced (Figures 4a and b), indicating an activation of the ERK pathway and inactivation of GSK3β. These data were also confirmed in NCCIT cells (Figure 4c). Interestingly, unlike the finding that SMAD2/3 activity was elevated when Akt activity was lost,13 we did not observe a significant increase in phosphorylation of SMAD2/3 in shCDK1- or RO3306-treated cells (Figures 4a–c). As a consequence of the CDK1-PDK1-PI3K/Akt-mediated alteration of ERK and GSK3β activities, the addition of a MEK/ERK inhibitor (UO126) blocked the upregulation of differentiation markers BRA, EOMES, and GSC and the downregulation of pluripotency markers NANOG, OCT4, and SOX2 (Figures 4d and e). The CDK1 inhibition-mediated upregulation of differentiation markers was also reversed with a SMAD2/3 inhibitor (SB431542) (Figure 4e). Convincingly, the blockage of differentiation by UO126 occurred by suppressing ERK activity, thereby releasing its inhibition of GSK3β (Figure 4f). Furthermore, the application of the PDK1 activator PS4824 partially reversed the CDK1 inhibition-mediated differentiation of hESCs (Figure 4g) through regulating ERK and GSK3β, the effectors of the PI3K/Akt signal pathway (Figure 4h). Collectively, these results demonstrate that CDK1-PDK1-Akt is a functional signaling pathway in controlling the pluripotency of hESCs via the suppression of ERK and maintenance of GSK3β activity.

Functional CDK1-PDK1-Akt pathway. (a) Immunoblot analysis of phosphorylated ERK, GSK3β, SMAD2/3, and their total proteins of the effectors of PI3K/Akt signaling in shControl- and shCDK1-transduced hESCs. (b) Immunoblot analysis of the activity of ERK, GSK3β, and SMAD2/3 in RO3306-treated hESCs. (c) Same analysis as (b) in NCCIT cells. (d) MEK/ERK inhibitor blocks CDK1 inactivation-mediated differentiation. hESCs were transduced with shControl and shCDK1±UO126 (5 μM), and the mRNA of mesoendoderm markers (left panel) and pluripotency marker (right panel) were analyzed by qRT-PCR. Paired Student’s t-test was used to compare between shCtrl and shCDK1; shCDK1 and shCDK1+UO126. *P<0.05, **P<0.01, ***P<0.0001. (e) hESCs were treated with DMSO (Ctrl), RO3306, RO3306+UO126, or RO3306+SB431542. The mRNA of mesoendoderm markers were analyzed by qRT-PCR. A statistical comparison was made between Ctrl and RO3306; RO3306 and RO3306+UO126; RO3306 and RO3306+SB431542. (f) Immunoblot analysis of pERK, pGSK3β, pSMAD2/3, and their total proteins in hESCs that were treated with RO3306 or RO3306+UO126. (g) hESCs were treated with RO3306 or RO3306+PS48 (3 μM), and the mRNA levels of OCT4 (left panel) and the mesoendoderm markers EOMES and GSC (right panel) were analyzed by qRT-PCR. (h) Immunoblot analysis of pERK, pGSK3β, pSMAD2/3, and their total proteins in hESCs that were treated with RO3306 or RO3306+PS48
Downregulation of CDK1 reduced metabolic glycolysis
High glycolytic rates have been characterized as metabolic requirements for the maintenance of pluripotency in many types of stem cells.25, 26 To explore whether CDK1-PDK1-Akt kinase pathway could modulate metabolic glycolysis in hESCs, which might further support the hypothesis of CDK1’s direct role in monitoring pluripotency, other than by regulating the cell cycle and proliferation. In this connection, it is known that inhibition of glycolysis does not affect cell proliferation.24 In CDK1-inactivated hESCs, expression of key glycolytic genes (GLUT1, PFK1, HK2, and LDHA) decreased significantly, as did lactate production in the culture medium (Supplementary Figures S6a and b). The decrease in expression of glycolysis genes was blocked after ERK suppression (Supplementary Figure S6c) and PDK1 activation (Supplementary Figure S6d). Thus, CDK1 inactivation-mediated hESC differentiation occurs via the inhibition of the PDK1/Akt signaling pathway and enhances the effector ERK activity, all of which are involved with glycolysis regulation.
Cyclin B1-CDK1 complex promotes somatic reprogramming
Given the role of CDK1 in maintaining hESC identity, we investigated whether CDK1 and its activator cyclin B1 have a role during somatic reprogramming. We observed a significant increase in the reprogramming efficiency of human fibroblasts after expressing cyclin B1 or co-expressing cyclin B1 with CDK1. The expression of CDK1 alone did not facilitate reprogramming. Furthermore, knocking down CDK1 in the background of cyclin B1 overexpression resulted in no induced pluripotent stem cells (iPSC) formation (Figure 5a and Supplementary Figure S7a). These results suggest that the improvement of iPSC efficiency by cyclin B1 depends on cyclin B1-CDK1 complexes. Similarly, expression of cyclin B1 promoted reprogramming efficiency in liver cancer epithelial cells (Figure 5b). The proportion of alkaline phosphatase(+) iPS colonies was significantly higher after ectopic expression of cyclin B1 (Figures 5c and d).

Cyclin B1-CDK1 complex promote somatic reprogramming and regulates LIN28A for iPSC maturation. (a) Human fibroblast cells were pre-infected with lentiviral empty vector, cyclin B1, CDK1, cyclin B1+CDK1, or cyclin B1+shCDK1, followed by iPS episomal transfection and reprogramming induction. The generating efficiency of iPSCs (%) was calculated by iPSC colonies produced versus cells seeded. (b) The iPSC generation efficiency in liver cancer cells 97 l that were transfected with lentiviral empty vector or cyclin B1. (c) Alkaline phosphatase(+) iPSC colonies versus total colonies after iPSC replating. *P<0.05. (d) Morphology of AP(+) colony comparison between vector-(upper panel) and cyclin B1-derived (lower panel) iPSC colonies. (e) Flow cytometry quantification of OCT4 and SSEA4 (left panel) and NANOG and TRA-1-60 (right panel) in nascent and replating iPS colonies. (f) Immunoblotting comparison of pluripotency proteins between vector- and cyclin B1-derived iPSCs nascent (left panel) and after replating (right panel). (g) mRNA comparison of NANOG, endogenous (Endo)-OCT4, -SOX2, and -LIN28A between vector- and cyclin B1-expressing iPSCs after replating; n=2 sets of iPSCs. (h) Morphology of cyclin B1-expressing iPSCs generated by iPS factors with and without LIN28. (i) mRNA comparison of NANOG, endo-OCT4, and -SOX2 in cyclin B1-expressing iPSC by iPS factors with and without LIN28. (j) mRNA comparison of NANOG, endo-OCT4, and -SOX2 in nascent and replating cyclin B1-expressing iPSCs by iPS factors without LIN28. (k) mRNA comparison of LIN28A and endo-LIN28A in nascent- and replating cyclin B1-expressing iPSCs by iPS factors without LIN28. AP, alkaline phosphatase
Cyclin B1 upregulated LIN28A for iPSC maturation
To explore the mechanism, pluripotency gene expression between nascent and replating reprogrammed cells were compared, as the pluripotency of iPSCs could be lost after replating.27 OCT4 and SSEA4 were expressed to similar levels in both vector- and cyclin B1-expressing iPSCs from the states of nascent to replating. Interestingly, the expression of NANOG and TRA-1-60, one of the best human pluripotency markers,27, 28 was higher in cyclin B1-expressing replating iPSCs than in the control cells (Figure 5e and Supplementary Figure S7b). The western blot of replating iPSCs displayed a similar result (Figure 5f). Cyclin B1 expression seems to have no notable effect on the cell cycle or proliferation in nascent or replating iPSCs (Supplementary Figure S7c). Only a small portion of initially formed iPSCs completed the reprogramming process and became iPSCs, whereas most of the iPSCs transitioned from TRA-1-60(+) into TRA-1-60(−) cells.27 Thus, in addition to enhancing reprogramming efficiency, cyclin B1 expression may also have a role in maintaining pluripotency after replating. We further found that after reprogramming with OCT4, SOX2, KLF4, L-MYC (OSKM), LIN28, and p53 shRNA,29 the expression of NANOG and endogenous OCT4 and SOX2 was comparable, whereas endogenous LIN28A level was significantly increased in cyclin B1-expressing iPSCs (Figure 5g), which might contribute to the maintenance of pluripotency after replating.
We then tried to generate iPSCs in the presence of cyclin B1 expression but without LIN28 (as well as without L-MYC).29 Indeed, iPSC colonies could be produced by the factors OCT4, SOX2, KLF4, and p53 shRNA without L-MYC or LIN28 (Figure 5h and Supplementary Figure S7d). Using this alternative iPSC system, the levels of NANOG, endogenous OCT4, and SOX2 were similar in iPSCs derived with iPSC factors with and without LIN28 (Figure 5i). iPSC colonies without LIN28 remained in undifferentiated states (Figure 5h) as well as with NANOG, and endogenous OCT4 and SOX2 expression remained high after replating (Figure 5j). Importantly, LIN28A and endogenous LIN28A expression was significantly increased after replating in these iPSCs (Figure 5k), indicating that cyclin B1-CDK1 complexes can upregulate and maintain cellular LIN28 expression, which is critical for iPSC maturation.27
Discussion
We demonstrated that CDK1 was enriched in pluripotent hESCs and was downregulated during differentiation; and there was an integrated correlation between the expression of pluripotency genes and CDK1. Downregulation of CDK1 activity could be manipulated at a level that did not disturb the cell cycle but still induced differentiation. Together, with the fact that NANOG levels were similar in different cell cycle phases during CDK1 inactivation-mediated differentiation, we propose that the link between ESC cell cycle and cell fate is not necessarily a consequence of cell cycle alteration, but rather occurs via a direct role of a cell cycle regulator in controlling pluripotency. Previous studies have focused on how cell cycle re-distribution leads to differentiation,30, 31, 32, 33, 34 although accumulating evidence suggests the direct control of pluripotency by cell cycle regulators.35, 36 Here, our study adds specific role of CDK1 in self-renewal and differentiation of hESCs beyond cell cycle regulation.
We demonstrate that (1) PDK1 is a putative downstream kinase of CDK1 as PDK1 contains an evolutionarily conserved CDK1 consensus phosphorylation site at Thr354; (2) CDK1 interacts with PDK1 and CDK1 kinase catalyzes phosphate transfer on PDK1 Thr354 peptide in vitro. Although we were not able to demonstrate specific phosphorylation on Thr354 in vivo, the fact that specific suppression of PDK1 phosphorylation and, accordingly, inhibited Akt phosphorylation by PDK1 when CDK1 was inactivated, indicates that PDK1 contains the specific activating site(s) for CDK1. Note that instead of PDK1, CDK1 knockdown may also have an impact on other kinases in affecting Akt activity, as PDK1 is not a direct kinase of Akt (Ser473). Thus, the inhibition of PDK1-Akt signaling pathway is responsible for the differentiation triggered by the inactivation of CDK1. The evidence that inhibition of Akt effectors blocked differentiation further indicates that the CDK1-PDK1-Akt signaling pathway is a functional pathway for pluripotency of hESCs via suppression of ERK and maintenance of GSK3β activity. Importantly, in addition to growth factors, which consistently stimulate and maintain high Akt signaling for self-renewal,13 CDK1 can regulate the essential PDK1-Akt signaling pathway for self-renewal, implicating a new kinase pathway in stem cell biology and the potential of chemical compounds that selectively decrease the level of CDK1 activity without perturbing cell cycle and proliferation for directing differentiation.
Interphase cyclin/CDKs are known to promote somatic reprogramming via increasing the rate of S phase cells.33, 34 We are the first to identify that mitotic driver cyclin B1-CDK1 complexes can increase efficiency of somatic reprogramming, which is unlikely through promoting cellular proliferation because co-expression of cyclin B1 with higher level of CDK1 inhibited iPSC formation (data not shown). Among the three known factors, LIN28, cyclin D1, and p53 shRNA that promote reprogramming activities, only LIN28 is considered a key regulator for iPSC maturation through inhibition of reprogramming reversion by enhancing TRA-1-60(+) proliferation and suppressing the conversion of TRA-1-60(+) to TRA-1-60(−) iPSCs, whereas cyclin D1 and p53 shRNA mainly promote cellular proliferation and suppress cell death.27 Here we found that cyclin B1-expressing iPSCs displayed a significantly high level of endogenous LIN28A exposed to iPS factors with or without exogenously added LIN28A. Apparently, cyclin B1 is able to upregulate and maintain cellular levels of LIN28A during reprogramming. Thus, we raise the possibility that monitoring iPSC factors could be a new path for improving reprogramming efficiency. In addition, p53 expression represses transcription of cyclin B1 and other mitotic regulators.37, 38 Application of p53 shRNA for reprogramming releases the repression and may further benefit reprogramming by cyclin B1 and CDK1.
Cancer cells are known to be refractory to reprogramming.39 Liver cancer cells contain a relatively higher level of LIN28.40, 41, 42 Under cyclin B1 expression, iPSC colonies can be successfully generated from liver cancer cells by iPS factors without LIN28A and L-MYC, suggesting that LIN28A is not a critical refractory factor to reprogramming. But enhancement of cellular LIN28A by cyclin B1 can overcome the resistance. Further study is needed to understand the mechanism how cyclin B1-CDK1 regulates LIN28A or other components for reprogramming. Recently, G2/M cell cycle regulators have been implicated in maintenance of pluripotency,43 where cyclin B1-CDK1 promotes iPSC maturation and provides new evidence from the point of view of somatic reprogramming.
Together, CDK1 is required for self-renewal of hESCs. The reduction of CDK1 activity to a level that does not disturb ESC cell cycling is able to suppress essential PDK1-PI3K/Akt signaling pathway and promote differentiation (Figure 6a and b). The sensitivity of hESCs to PI3K/Akt signaling can be further regulated by the CDK1-PDK1-PI3K/Akt kinase cascade (Figure 6b). Cyclin B1-CDK1 complexes are important during reprogramming, probably via regulating cellular LIN28A for iPSC maturation. This study provides a novel kinase cascade mechanism for pluripotency control and acquisition.

A proposed function of CDK1 in regulating pluripotency. (a) The correlation of CDK1 activity with embryonic development, cell cycle, and self-renewal/differentiation. (b) Model of the CDK1-PDK1-PI3K/Akt kinase pathway for self-renewal and differentiation. CDK1 maintains phosphorylation of PDK1 and hyperactive Akt (indicated by +) for self-renewal via inhibiting pERK and maintaining GSK3β activity. Upon the reduction of CDK1 activity, the phosphorylation of PDK1 and Akt is sequentially suppressed (indicated by −); ERK inhibition is relieved to suppress GSK3β kinase, which results in stabilization of β-catenin. Subsequently, pSMAD2/3 and Wnt effectors cooperate to promote differentiation
Materials and Methods
Cell culture
The hESC lines H1, H7, and H9 were maintained in a feeder-free mTeSR1 medium (Stemcell Technologies, Vancouver, Canada) on Matrigel (BD Biosciences, San Jose, CA, USA) or Geltrex (Life Technologies, Grand Island, NY, USA). NCCIT cells (human pluripotent embryonal carcinoma line) were cultured according to ATCC’s instructions. Lentivirus particles were produced in 293 T cells using ViraPower Lentiviral Packaging Mix (Life Technologies) and concentrated by ultracentrifugation (20 000 × g). hESCs were infected with 1 ml of shRNA lentivirus for 1 h and then cultured in 50% lentiviral and 50% mTeSR1 medium for 18 h followed by changing into full mTeSR1 medium to recover.34 hESCs were treated with CDK1 inhibitor RO3306 (Merck, Kenilworth, NJ, USA) at a dose of 5–8 μM for 2–3 days before further experiments. Other chemical compounds that were used include UO126, SB431542 (Merck), PS48 (Stemgent, Cambridge, MA, USA), and JNJ-7706621 (Selleckchem, Houston, TX, USA). DMSO was applied as a vehicle control. In vitro differentiation of hESC was induced via (1) EB differentiation: the formation of EBs followed by differentiation in normal growth medium without βFGF44 (KSR medium); (2) FBS differentiation: EBs were further cultured in DMEM+20% FBS; and (3) retinoic acid (Sigma-Aldrich, St. Louis, MI, USA) differentiation: 2–5 μM of retinoic acid was added to hESC or NCCIT cell culture.
Vectors for knockdown and overexpression
Plasmids expressing shRNAs against human CDK1, NANOG, and OCT4 were constructed using the pEco-Lenti-H1-shRNA (GFP) kit (GenTarget Inc., San Diego, CA, USA). The oligos for shRNAs were designed by Life Technologies RNAi Designer or Gene Link shRNA design program and are listed in Supplementary Information Supplementary Table S1. Human cyclin B1 and CDK1 genes from hESCs were cloned into pWPI-lentiviral vector. Human iPSC constructs, including three episomal plasmid vectors (pCXLE-hOCT3/4-shp53, pCXLE-hSK, and pCXLE-UL) encoding human OCT4, SOX2, KLF4, LIN28, L-MYC, and p53 shRNA29 were from Addgene.
qRT-PCR
Total RNA isolation, DNase I treatment, and reverse transcription were performed as previously described.45 Quantitative PCR was performed using the SYBR Green master mix (Life Technologies) on 7900HT Detection System with primers listed in Supplementary Information Supplementary Table S1. All the PCRs were repeated 2–3 times with each run in duplicate and normalized to 18S rRNA. Relative quantification was calculated as ΔΔCT, which is relative to cells that were treated with scramble shRNA or vehicle.
Cell cycle analysis and flow cytometry
Cell cycle analysis was performed using a BrdU and propidium iodide staining assay after fixation as previously described.46 Cells were stained with antibodies for pluripotency markers and analyzed using FACSCalibur (BD Biosciences). The antibodies included NANOG, OCT4, SOX2 (Cell Signaling Technology, Danvers, MA, USA), SSEA4 (BD Biosciences), and TRA-1-60 (Cell Signaling Technology; Biosource, CA, USA). The MitoProbeDilC1(5) assay kit (Life Technologies) was used to detect live, apoptotic, and dead cell populations in one staining reaction.
Immunoblot and co-immunoprecipitation
Cell lysates for the western blots were prepared as described previously.46 Membranes were probed with the following antibodies: β-actin (Sigma-Aldrich); CDK1 and OCT4 (Santa Cruz Biotechnology, Dallas, TX, USA); CDK2 (Abcam, Bristol, UK); and phospho-Akt (Thr308, Ser473), phosphor-ERK1/2 (Thr202/Tyr204), phospho-SMAD2/3 (pSMAD2-Ser465/467, pSMAD3-Ser423/425), phospho-GSK3β (Ser9), phospho-PDK1 (Ser241), NANOG, OCT4, and SOX2 (Cell Signaling Technology). Co-immunoprecipitation was performed using phospho- (Ser/Thr) PDK1 docking motif antibodies and phospho-Akt antibodies. Total cell lysates (100–200 μg) were pre-cleaned and incubated with phospho-(Ser/Thr) PDK1 docking motif antibody (Cell Signaling Technology) overnight, before incubated with 50% bead slurry for 2 h. After five washes, the immune complexes were analyzed by immunoblotting with anti-phospho-Akt antibodies. Co-immunoprecipitation for assessing interaction was carried out using CDK1 antibody for immunoprecipitation; the bound proteins were analyzed by immunoblotting with PDK1 antibody.
Kinase assay
Total cell lysates (200 μg) were pre-cleaned with blank beads and incubated with CDK1 antibody overnight followed by incubation with 50% protein G bead slurry for 3 h. After five washes (the last two washes using kinase buffer), the immune complexes were incubated with peptide substrates PDK1 Thr354-WT (ENLHQQTPPKLTA) or Thr354A mutant (ENLHQQAPPKLTA) along with kinase reaction mix from ADPsensor universal kinase activity assay kit (BioVision, Milpitas, CA, USA) following the manufacturer’s instructions. Fluorescent signal at Ex/Em=535/587 was read using Microplate Fluorescence Reader FL600 (BIO-TEX Instruments, Winooski, VT, USA).
Colony-forming assays and alkaline phosphatase staining
The AP staining kit (Millipore, Ontario, Canada; Sigma-Aldrich) was used to differentially stain pluripotent colonies before the number of colonies was quantified.
Lactate production measurement
Lactate production in the culture medium of hESCs was measured using the Lactate Assay Kit (BioVision) after RO3306 and other chemical treatments. The hESC cultures that were treated with vehicles or various chemicals were maintained without changing the medium for 2 days to accumulate lactate.
Somatic cells reprogramming
Pre-infected lentiviral-cyclin B1, CDK1, or shCDK1 human diploid fibroblasts or liver cancer cells (97 L) were transfected with equal amount of iPS episomal vectors using electroporation (Lonza, Basel, Switzerland) followed by passage onto inactivated MEF and switched to iPSC medium 5–6 days later.29 iPS colony formation was observed and compared between 14 days and 20 days. The pluripotency of iPSC colonies were stained for alkaline phosphatase. The reprogramming frequency was calculated in the percentage of the number of iPS colonies that were generated versus overall cells that were seeded on MEF. From the first formed iPSCs, several colonies were randomly picked and passed on to fresh feeder cells for replating. After replating on MEF, the iPSC colonies were transferred to Geltrex-coated dishes. iPSC colonies were also generated under feeder-free conditions using ReproTeSR medium (Stemcell Technologies) following the manufacturer’s instruction.
Statistical analysis
All data were represented as the mean±S.D. Student’s t-test (paired or independent t-test) was performed between the controls and experimental groups using SPSS 21 (IBM Corp., Armonk, NY, USA). A P-value <0.05 was considered statistically significant.
Abbreviations
- hESCs:
-
human Embryonic Stem Cells
- iPSCs:
-
induced Pluripotency Stem Cells
- EB:
-
embryoid body
- RO:
-
RO3306
- JNJ:
-
JNJ-770621
- UO:
-
UO126
- SB:
-
SB431542
- OSKM:
-
OCT4, SOX2, KLF4, L-MYC
- PDK1:
-
phosphoinositide-dependent kinase-1
References
Hu X, Moscinski LC . Cdc2: a monopotent or pluripotent CDK? Cell Prolif 2011; 44: 205–211.
Santamaría D, Barrière C, Cerqueira A, Hunt S, Tardyk C, Newton K et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007; 448: 811–815.
Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci USA 2012; 109: 3826–3831.
Adhikari D, Zheng W, Shen Y, Gorre N, Ning Y, Halet G et al. Cdk1, but not Cdk2, is the sole Cdk that is essential and sufficient to drive resumption of meiosis in mouse oocytes. Hum Mol Genet 2012; 21: 2476–2484.
Ullah Z, Kohn MJ, Yagi R, Vassilev LT, DePamphilis ML . Differentiation of trophoblast stem cells into giant cells is triggered by p57/Kip2 inhibition of CDK1 activity. Genes Dev 2008; 22: 3024–3036.
Zhang WW, Zhang XJ, Liu HX, Chen J, Ren YH, Huang DG et al. Cdk1 is required for the self-renewal of mouse embryonic stem cells. J Cell Biochem 2011; 112: 942–948.
Neganova I, Tilgner K, Buskin A, Paraskevopoulou I, Atkinson SP, Peberdy D et al. CDK1 plays an important role in the maintenance of pluripotency and genomic stability in human pluripotent stem cells. Cell Death Dis 2014; 5: e1508.
Li L, Wang J, Hou J, Wu Z, Zhuang Y, Lu M et al. Cdk1 interplays with Oct4 to repress differentiation of embryonic stem cells into trophectoderm. FEBS Lett 2012; 586: 4100–4107.
Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW et al. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006; 444: 364–368.
Van Hoof D, Muñoz J, Braam SR, Pinkse WH, Linding R, Heck JR et al. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell 2009; 5: 214–226.
Ng HH, Surani MA . The transcriptional and signaling networks of pluripotency. Nat Cell Biol 2011; 13: 490–496.
Nichols J, Smith A . Naïve and primed pluripotent states. Cell Stem Cell 2009; 4: 487–492.
Singh AM, Reynolds D, Cliff T, Ohtsuka S, Mattheyses AL, Sun YH et al. Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 2012; 10: 312–326.
Oshimon N, Fuchs E . The harmonies played by TGF-β in stem cell biology. Cell Stem Cell 2012; 11: 751–764.
Chen YG, Li Z, Wang XF . Where PI3K/Akt meets Smads: the crosstalk determines human embryonic stem cell fate. Cell Stem Cell 2012; 10: 231–232.
Dutta D . Signaling pathways dictating pluripotency in embryonic stem cells. Int J Dev Biol 2013; 57: 667–675.
Singh AM, Bechard M, Smith K, Dalton S . Reconciling the different roles of Gsk3β in “naïve” and “primed” pluripotent stem cells. Cell Cycle 2012; 11: 2991–2996.
Pearce LR, Komander D, Alessi DR . The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010; 11: 9–22.
Casamayor A, Morrice NA, Alessi DR . Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo . Biochem J 1999; 342: 287–292.
Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao XL, Sun H et al. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci USA 2006; 103: 10660–10665.
Emanuel S, Rugg CA, Gruninger RH, Lin R, Fuentes-Pesquera A, Connolly PJ et al. The in vitro and in vivo effects of JNJ-7706621: a dual inhibitor of cyclin-dependent kinases and aurora kinases. Cancer Res 2005; 65: 9038–9046.
Hill MM, Andjelkovic M, Brazil DP, Ferrari S, Fabbro D, Hemmings BA . Insulin-stimulated protein kinase B phosphorylation on Ser-473 is independent of its activity and occurs through a staurosporine-insensitive kinase. J Biol Chem 2001; 276: 25643–25646.
Song G, Ouyang G, Bao S . The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 2005; 9: 59–71.
Zhu S, Li W, Zhou H, Wei WG, Ambasudhan R, Lin TX et al. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010; 7: 651–655.
Ito K, Suda T . Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol 2014; 15: 243–256.
Ward PS, Thompson CB . Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol 2012; 4: a006783.
Tanabe K, Nakamura M, Narita M, Takahashi K, Yamanaka S . Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proc Natl Acad Sci USA 2013; 110: 12172–12179.
Chan EM, Ratanasirintrawoot S, Park IH, Manos PD, Loh YH, Huo H et al. Live cell imaging distinguishes bonafide human iPS cells from partially reprogrammed cells. Nat Biotechnol 2009; 27: 1033–1037.
Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S et al. A more efficient method to generate integration-free human iPS cells. Nat Methods 2011; 8: 409–412.
Pauklin S, Ludovic Vallier L . The cell-cycle state of stem cells determines cell fate propensity. Cell 2013; 155: 135–147.
Momcilović O, Choi S, Varum S, Bakkenist C, Schatten G, Navara C . Ionizing radiation induces ataxia telangiectasia mutated-dependent checkpoint signaling and G(2) but not G(1) cell cycle arrest in pluripotent human embryonic stem cells. Stem Cells 2009; 27: 1822–1835.
Conklin JF, Baker J, Sage J . The RB family is required for the self-renewal and survival of human embryonic stem cells. Nat Commun 2012; 3: 1244.
Ruiz S, Panopoulos AD, Herrerías A, Bissig KD, Lutz M, Berggren WT et al. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr Biol 2011; 21: 45–52.
Edel MJ, Menchon C, Menendez S, Consiglio A, Raya A, Izpisua Belmonte JC . Rem2 GTPase maintains survival of human embryonic stem cells as well as enhancing reprogramming by regulating p53 and cyclin D1. Genes Dev 2010; 24: 561–573.
Lee DF, Su J, Ang YS, Carvajal-Vergara X, Mulero-Navarro S, Pereira CF et al. Regulation of embryonic and induced pluripotency by aurora kinase-p53 signaling. Cell Stem Cell 2012; 11: 179–194.
Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015; 16: 39–50.
Taylor WR, DePrimo SE, Agarwal A, Agarwal ML, Schönthal AH, Katula KS et al. Mechanisms of G2 arrest in response to overexpression of p53. Mol Biol Cell 1999; 10: 3607–3622.
Innocente SA, Lee JM . p53 is a NF-Y- and p21-independent, Sp1-dependent repressor of cyclin B1 transcription. FEBS Lett 2005; 579: 1001–1007.
Kim J, Zaret KS . Reprogramming of human cancer cells to pluripotency for models of cancer progression. EMBO J 2015; 34: 739–747.
Tian N, Han Z, Li Z, Zhou M, Fan C . Lin28/let-7/Bcl-xL pathway: the underlying mechanism of drug resistance in Hep3B cells. Oncol Rep 2014; 32: 1050–1056.
Zheng YW, Nie YZ, Taniguchi H . Cellular reprogramming and hepatocellular carcinoma development. World J Gastroenterol 2013; 19: 8850–8860.
Yin X, Li YW, Jin JJ, Zhou Y, Ren ZG, Qiu SJ et al. The clinical and prognostic implications of pluripotent stem cell gene expression in hepatocellular carcinoma. Oncol Lett 2013; 5: 1155–1162.
Gonzales KA, Liang H, Lim YS, Chan YS, Yeo JC, Tan CP et al. Deterministic restriction on pluripotent state dissolution by cell-cycle pathways. Cell 2015; 162: 564–579.
Braam SR, Denning C, Matsa E, Young LE, Passier R, Mummery CL . Feeder-free culture of human embryonic stem cells in conditioned medium for efficient genetic modification. Nat Protoc 2008; 3: 1435–1443.
Wang XQ, Ng RK, Ming X, Zhang W, Chen L, Chu AC et al. Epigenetic regulation of pluripotent genes mediates stem cell features in human hepatocellular carcinoma and cancer cell lines. PLos One 2013; 8: e72435.
Wang X, Lui VC, Poon RT, Lu P, Poon RY . DNA damage mediated s and g(2) checkpoints in human embryonal carcinoma cells. Stem Cells 2009; 27: 568–576.
Acknowledgements
We thank Professor Chris Norbury (University of Oxford) for his critical suggestions for the experiments design; Dr. Ray Ng (The University of Hong Kong) for providing lentiviral vector; Professor Eric Stanbridge (University of California Irvine) for his advice and language editing. This work was supported by the Seed Funding Program for Basic Research, The University of Hong Kong and Health and Medical Research Fund Research Council of Hong Kong (59190, 59047, 59050, and 01122246 to XQW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by R De Maria
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Wang, X., Lo, C., Chen, L. et al. CDK1-PDK1-PI3K/Akt signaling pathway regulates embryonic and induced pluripotency. Cell Death Differ 24, 38–48 (2017). https://doi.org/10.1038/cdd.2016.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.84
This article is cited by
-
RACGAP1 promotes the progression and poor prognosis of lung adenocarcinoma through its effects on the cell cycle and tumor stemness
BMC Cancer (2024)
-
SPINK1-induced tumor plasticity provides a therapeutic window for chemotherapy in hepatocellular carcinoma
Nature Communications (2023)
-
BEX1 supports the stemness of hepatoblastoma by facilitating Warburg effect in a PPARγ/PDK1 dependent manner
British Journal of Cancer (2023)
-
Macrophages maintain mammary stem cell activity and mammary homeostasis via TNF-α-PI3K-Cdk1/Cyclin B1 axis
npj Regenerative Medicine (2023)
-
iPSCs derived from insulin resistant offspring of type 2 diabetic patients show increased oxidative stress and lactate secretion
Stem Cell Research & Therapy (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}