
Abstract
The AMP-activated protein kinase, a key regulator of energy homeostasis, has a critical role in metabolic disorders and cancers. AMPK is mainly regulated by cellular AMP and phosphorylation by upstream kinases. Here, we show that PIKE-A binds to AMPK and blocks its tumor suppressive actions, which are mediated by tyrosine kinase Fyn. PIKE-A directly interacts with AMPK catalytic alpha subunit and impairs T172 phosphorylation, leading to repression of its kinase activity on the downstream targets. Mutation of Fyn phosphorylation sites on PIKE-A, depletion of Fyn, or pharmacological inhibition of Fyn blunts the association between PIKE-A and AMPK, resulting in loss of its inhibitory effect on AMPK. Cell proliferation and oncogenic assays demonstrate that PIKE-A antagonizes tumor suppressive actions of AMPK. In human glioblastoma samples, PIKE-A expression inversely correlates with the p-AMPK levels, supporting that PIKE-A negatively regulates AMPK activity in cancers. Thus, our findings provide additional layer of molecular regulation of the AMPK signaling pathway in cancer progression.
Similar content being viewed by others


Main
AMP-activated protein kinase is activated under a variety of physiological and pathological stresses that increase the intracellular AMP/ATP ratio, either by increasing ATP consumption (exercise/muscle contraction) or by decreasing ATP production (e.g., glucose deprivation, hypoxia or ischemia). It is a heterotrimeric complex consisting of a catalytic α subunit and two regulatory (β and γ) subunits. An increase in intracellular AMP/ATP ratio results in allosteric activation of the kinase by protecting T172 from dephosphorylation.1 T172 phosphorylation in the activation loop of the α subunit is an absolute requirement for full activation of AMPK activity,2, 3 and is mediated by at least two distinct upstream kinases, liver kinase B1 (LKB1)4, 5, 6 and Ca2+/calmodulin-dependent kinase kinase β (CaMKKβ).7, 8, 9
AMPK is an evolutionarily conserved metabolic sensor that has a pivotal role in maintaining energy homeostasis by coordinating metabolic pathways to balance nutrient supply and demand.10 Regulation of AMPK in multiple tissues is controlled by a growing number of hormones and cytokines, including leptin, adiponectin, IL-6, CNTF, TNF-α, and ghrelin. Moreover, AMPK can be activated by numerous small molecules such as metformin, aminoimidazole-4-carboxymide-1-β-D-ribofuranoside (AICAR), resveratrol, thiozolidinedione (TZD), and A-769662. Activated AMPK regulates glucose uptake and fatty acid oxidation in muscle and blocks gluconeogenesis in liver, enhancing insulin sensitivity. It also regulate appetite (for review, see Dzamko and Steinberg).11 In addition to these well-characterized functions in metabolic syndromes, AMPK serves as a metabolic tumor suppressor that reprograms the cellular metabolism and elicits a metabolic checkpoint on the cell cycle through its actions on mTORC1, p53, and other modulators for cell proliferation, cell growth, cell survival, and autophagy.12 Further, LKB1 activates AMPK and represses RNA synthesis.13 In LKB1-deficient lung cancer cells, AMPK activity is suppressed, leading to increased cell growth, whereas the ability of AMPK to inhibit cell growth is restored when wild-type LKB1 is expressed.14, 15 Additionally, the express levels of AMPK inversely correlate with clinical prognosis in gastric,16 breast, and ovarian tumors, and are diminished in cancer cells by activated PI3K pathways.17 Accumulating evidence supports that the susceptibility of cancer might be attributable to the dysregulated AMPK.18, 19 Hence, activation of AMPK may represent a novel target for cancer treatment.
PIKE-A is a GTPase that directly interacts with PI 3 kinase or Akt and enhances their kinase activities.20, 21, 22, 23 It is a proto-oncogene that frequently amplified in numerous human cancers.24, 25 It binds Akt and escalates its kinase activity and promotes cancer cell survival, invasion, and migration.26, 27 Interestingly, PIKE knockout (PIKE−/−) mice are resistant to diet-induced obesity and diabetes,28 strongly implicating PIKE in obesity control. Accordingly, we observed higher AMPK phosphorylation and lipid oxidation in PIKE−/− muscle and fat tissues, which provide a mechanistic explanation to the slim phenotype of the knockout mice.28 Further, PIKE-A interacts with insulin receptor and mediates its suppressive effect on AMPK activation.29 Previously, we have reported that Fyn phosphorylates PIKE-A on both Y682 and Y77430 and regulates its interaction with different partners, promoting neuronal survival31 and adiposeness.32 In this report, we provide new evidence supporting that Fyn phosphorylation of PIKE-A is critical for its association with AMPK and inhibition of its kinase activity, leading to the blockade of cell proliferation. Hence, PIKE-A promotes tumorigenesis, at least, partially through blocking the tumor suppressive activity of AMPK. This discovery highlights a previously unappreciated relationship between cell metabolism and cell proliferation mediated by PIKE-A/AMPK complex.
Results
PIKE-A directly associates with AMPK
Knockout of PIKE-A leads to upregulation of AMPK activity in muscle and fat tissues, indicating that PIKE-A might negatively regulate AMPK signaling.28 A reciprocal immunoprecipitation in LN229 glioblastoma cells showed that endogenous PIKE-A specifically associated with AMPK (Figure 1a). In vitro binding assay also showed that PIKE-A directly interacted with AMPKα (Supplementary Figure S1d). GST pull-down experiment revealed that both α1 and 2 but not β or γ subunits of AMPK interacted with PIKE-A (Supplementary Figure S1a). Truncation experiment demonstrated that the AMPK α2 kinase domain was implicated in binding to PIKE-A (Figure 1b). On the other hand, deletion of GTPase domain in PIKE-A abolished the interaction (Figure 1c), indicating that the GTPase domain is required for PIKE-A to interact with AMPKα subunit. The binding assay validated that AMPKα directly interacted with PIKE-A GTPase domain (a.a. 73–340) (Figure 1d). AMPKα1 subunit also binds to PIKE-A GTPase domain (Supplementary Figure S1a), supporting that both α1 and α2 isoforms of AMPK directly interact with PIKE-A GTPase domain. To explore whether the kinase activity of AMPKα is required for its association with PIKE-A, we performed GST pull-down assay using kinase-dead AMPKα. Both wild-type (wt) AMPKα and AMPKα K45R mutant bound to PIKE-A (Supplementary Figures S1c and d). Hence, PIKE-A physically associates with AMPK in cancer cells.

PIKE-A directly associates with AMPK. (a) Reciprocal immunoprecipitation was conducted in human glioblastoma cell line LN229. In all, 2 mg of cell lysate was immunoprecipitated with anti-PIKE-A (or anti-AMPKα) and immunoblotted using anti-AMPKα (or anti-PIKE-A). In all, 60 μg of cell lysate was used as an input control. (b) GST-PIKE-A construct was cotransfected with various truncates of AMPKα into HEK293 cells. Cell lysates were pulled down with glutathione beads and analyzed by immunoblotting with anti-myc antibody. (c) Different PIKE-A truncates were cotransfected with Flag-tagged AMPKα1 into HEK293 cells, and PIKE-A fragments were immunoprecipitated with anti-myc antibody, and coprecipitated proteins were analyzed by immunoblotting with anti-Flag. (d) GST-tagged recombinant proteins of various PIKE-A domains were purified and incubated with cell lysates, transfected with myc-AMPKα2. Glutathione bead-associated proteins were analyzed by immunoblotting with anti-myc
PIKE-A suppresses AMPK signaling pathways
Transfection of PIKE-A wt and KS mutant, which binds to Akt but fails to upregulate its kinase activity,24, 26 into two glioblastoma cell lines LN229 and U87MG demonstrated that wt PIKE-A overexpression strongly repressed phosphorylations of AMPK and its downstream target acetyl Co-A carboxylase (ACC) (Figure 2a). Moreover, PIKE-A activated Akt, which subsequently stimulated mTOR activity, leading to an increase in p70S6K phosphorylation. AMPK arrests the cell cycle at G1 phase through increasing p53 phosphorylation, leading to the upregulation of p21.33 Further, AMPK activation also causes phosphorylation and stabilization of cell cycle-dependent kinase inhibitor p27kip1.34 Accordingly, we observed a reduction of p21 and p27, when PIKE-A was overexpressed. By contrast, when PIKE-A-KS was overexpressed, AMPK/ACC signaling was partially relieved, so were the downstream signalings of mTORC1 and p53 pathways. Fitting with previous observations,26 PIKE-A WT prominently increased Akt phosphorylation, whereas KS mutant lost its stimulatory effect (Figure 2a). Depletion of PIKE-A elicited an upregulation of AMPK/ACC pathway but reduced the phosphorylation of p70S6K and elevated p21 and p27. Akt phosphorylation was evidently blocked when PIKE-A was eliminated (Figure 2b).

PIKE-A inhibits AMPK signaling pathway. (a) LN229 and U87MG glioblastoma cell lines were transfected with GFP-PIKE-A or GTPase-dead PIKE-A-KS and AMPKα and its downstream effectors including ACC, p70S6K, p21, and p27 were analyzed by immunoblotting. (b) LN229 and U87MG cells were infected with shRNA of PIKE-A adenovirus, and AMPKα and its downstream effectors in the cell lysates were analyzed by immunoblotting. (c) HEK293 cells were transfected with GFP vector, GFP-PIKE-A, or GFP-PIKE-KS. The endogenous AMPK was immunoprecipitated and subjected to SAMS kinase assay. The AICAR was a positive control. Immunoblotting analysis was performed for p-AMPKα/p-ACC in the transfected cells (*P<0.05, **P<0.01, n=3)
To examine whether PIKE-A directly inhibits AMPK activity, we performed SAMS kinase assay employing immunoprecipitated AMPK complex from HEK293 cells transfected with control GFP, GFP-PIKE-A, or GFP-PIKE-A-KS. The peptide phosphorylation was significantly decreased when PIKE-A was overexpressed; in contrast, PIKE-A KS lost its inhibitory effect. In vitro SAMS kinase assay with active AMPK pre-incubated with purified GST, GST-PIKE-A or GST-PIKE-A KS recombinant proteins displayed the same result (Figure 2c and Supplementary Figure S2a). AMPK signaling was strongly activated in PIKE−/− mouse embryonic fibroblasts (MEFs) when compared with wt (PIKE+/+) MEF. AMPK signaling pathway was further escalated by the well-characterized stimuli, including H2O2, AICAR, metformin, and A23187 (Supplementary Figure S2b). Hence, these data strongly support that PIKE-A binds to AMPK and blocks its signaling pathways.
Fyn phosphorylation regulates the association between PIKE-A and AMPK
Fyn phosphorylates PIKE-A on Y682 and Y774.30 The tyrosine phosphorylation status on PIKE-A tightly correlated with the binding affinity by PIKE-A to AMPKα (Figure 3a). Notably, Fyn-CA (constitutively active) enhanced the binding between PIKE-A and AMPK, and Fyn-KD (kinase-dead) decreased the interaction. PIKE-A tyrosine phosphorylation was stronger with Fyn-CA than control or Fyn-KD expressed cells. Compared with the prominent interaction between PIKE-A-wt and AMPK, unphosphorylate PIKE-A mutant FF (Y682F, Y774F) lost its binding affinity to AMPK. Furthermore, Fyn-CA escalated the PIKE-A wt/AMPK association, but its stimulatory effect was reduced when PIKE-A-FF was employed. As expected, PIKE-A FF barely interacted with AMPKα in the presence of Fyn-KD (Figure 3a). To further confirm that Fyn phosphorylation of PIKE-A has a role in regulating the PIKE-A/AMPK complex formation, we utilized a Fyn tyrosine kinase inhibitor, PP2. In the presence of PP2, the association was blocked, whereas PP3, an inactive pharmacologic analog of PP2, had no effect (Figure 3b). Reciprocal immunoprecipitation using either anti-PIKE-A or anti-AMPKα antibodies in brain and muscle tissues showed that knockout of Fyn substantially dissociated the PIKE-A/AMPK complex (Figure 3c). These data support that Fyn tyrosine phosphorylation is required for the PIKE-A/AMPK complex formation.

Fyn phosphorylation regulates the association between PIKE-A and AMPK. (a) HEK293 cells were cotransfected with myc-AMPKα2 with GST-PIKE wild-type or unphosphorylate mutant PIKE-A-FF in the presence of HA-Fyn CA or HA-Fyn KD. PIKE-A was pulled down with glutathione beads and coprecipitated proteins were analyzed by immunoblotting with anti-myc or anti-PY20. The expression levels of transfected constructs were analyzed by immunoblotting. (b) HEK293 cells were cotransfected with GST-PIKE-A and myc-AMPKα2, followed by treatment with Fyn kinase inhibitor PP2 (1 μM) or its inactive analog PP3 (1 μM) for 24 h. PIKE-A was pulled down with glutathione beads, and coprecipitated proteins were analyzed by immunoblotting with anti-myc or anti-PY20 antibody. (c) The reciprocal immunoprecipitation was conducted with wild-type or Fyn−/− brain and muscle tissues. The expression of PIKE-A, AMPK, and Fyn was validated in these tissues
Fyn phosphorylation is required for PIKE-A to inhibit AMPK signaling pathways
Previous study reveals that Fyn inhibits AMPK signaling through phosphorylating LKB1, leading to its nuclear translocation.35 To test the biological effect of Fyn phosphorylation of PIKE-A in mediating AMPK signaling cascades, we employed LKB1-deficient A549 lung adenocarcinoma cells to avoid the contribution of Fyn/LKB1 in this event. Remarkably, Fyn-CA robustly inhibited AMPK/ACC signaling compared with mock control, whereas this inhibitory effect was impaired with Fyn-KD (Figure 4a). Overexpression of PIKE-A repressed AMPK/ACC signalings, and this repressive activity was further escalated in the presence of Fyn-CA. However, when Fyn-KD was cotransfected, the inhibitory activity of PIKE-A was abolished, indicating that phosphorylation by Fyn is essential for PIKE-A to block AMPK/ACC signaling. On the other hand, Fyn-CA was unable to suppress AMPK/ACC signaling in the presence of PIKE-A FF mutant, suggesting that Fyn exerts its inhibitory effect on AMPK pathway through phosphorylating PIKE-A. Noticeably, both PIKE-A wt and PIKE-A-FF failed to repress AMPK/ACC signaling in the presence of Fyn-KD, further indicating that PIKE-A phosphorylation by Fyn is indispensable for its suppressive activity (Figure 4a).

Fyn-phosphorylated PIKE-A inhibits AMPK signaling pathways. (a) A549 cells were transfected with Fyn-CA, Fyn-KD, GST-PIKE-A WT, or PIKE-A-FF. AMPK and ACC phosphorylation status and transfected constructs were analyzed by immunoblotting. (b) A549 cells were infected with shPIKE adenovirus, followed by Fyn-CA overexpression. AMPKα and ACC phosphorylation status was analyzed by immunoblotting. (c) PIKE-A +/+ and −/− MEFs were transfected with HA-Fyn-CA or HA-Fyn-KD. AMPKα and ACC phosphorylation status was analyzed by immunoblotting. (d and e) The wild-type or PIKE-A-null MEFs and HeLa cells were transfected with control siRNA or Fyn-specific siRNA. AMPKα/ACC phosphorylation signals and validation of Fyn knockdown by its siRNA were analyzed by immunoblotting
In A549 cells, Fyn-CA strongly blocked AMPK/ACC signaling. The inhibitory activity of Fyn-CA on AMPK/ACC signaling was significantly reduced when PIKE-A was depleted (Figure 4b and Supplementary Figure S3a). Whereas Fyn-CA suppressed AMPK/ACC signaling in PIKE+/+ MEF compared with Fyn-KD, its inhibitory effect was clearly reduced in PIKE−/− MEF (Figure 4c). Thus, these findings indicate that PIKE-A mediates the inhibitory actions of Fyn toward AMPK/ACC pathway. Depletion of Fyn also elevated AMPK/ACC signaling in wt MEF, which was further augmented in PIKE−/− MEF (Figure 4d), indicating that PIKE-A is involved in the Fyn-AMPK cascade. Strikingly, depletion of Fyn evidently upregulated AMPK/ACC signaling cascade in LKB1-deficient HeLa cells (Figure 4e), suggesting that LKB1 is not the only effector for Fyn to regulate AMPK activity. Collectively, our data support that PIKE-A directly interacts with AMPK and inhibits its kinase activity, which is regulated by Fyn phosphorylation on PIKE-A.
PIKE-A inhibits AMPK phosphorylation by LKB1 or CaMKKβ
To determine how PIKE-A antagonizes AMPK phosphorylation and activation, we chose LKB1-deficient A549 cells. Overexpression of LKB1 complex (LKB1/STRAD/MO25) elicited AMPK and ACC phosphorylation. By contrast, the kinase-dead LKB1 (K78M) reduced AMPK phosphorylation. Noticeably, LKB1’s stimulatory effect on phospho-AMPK was reduced by PIKE-A (Figure 5a). We made similar observations in LKB1-deficeint HeLa cells as well (data not shown). Accordingly, AMPK phosphorylation was substantially reduced in LKB1−/− MEF compared with wt MEF. Depletion of PIKE-A strongly escalated AMPK signalings in both wt and LKB1−/− MEFs (Figure 5b). These data indicate that PIKE-A may block AMPK phosphorylation by CaMKKβ in A549 cells or LKB−/− MEF cells. Notably, overexpression of PIKE-A wt but not unbound mutant FF decreased p-AMPK/p-ACC. PIKE-A wt resulted in complete inhibition of p-AMPKα, whereas PIKE-A-FF mutant failed in LKB1−/− MEF cells (Figure 5c). Hence, PIKE-A inhibits AMPK phosphorylation by both LKB1 and CaMKKβ. To directly test the inhibitory actions by PIKE-A on AMPK phosphorylation by LKB1, we performed an in vitro kinase assay employing the kinase-dead AMPKα2 K45R as a substrate in the presence of PIKE-A WT or FF recombinant proteins. Active LKB1 complex robustly triggered AMPKα2 K45R phosphorylation, which was inhibited by recombinant GST-PIKE-A but not FF mutant (Figure 5d). We also performed the same experiment with purified active CaMKKβ, and made a similar observation (Figure 5e). PIKE-A did not mediate the association between AMPKα and its upstream kinases, LKB1 and CaMKKβ, respectively (Supplementary Figure S3b). PIKE-A did not block myelin basic protein (MBP) peptide phosphorylation by LKB1 (Supplementary Figure S3c), indicating that PIKE-A does not directly inhibit LKB1’s kinase activity. In LKB1-deficient A549 cells, knocking down of CaMKKβ abolished AMPK phosphorylation (Supplementary Figure S3d), indicating that CaMKKβ is the major kinase responsible for AMPK phosphorylation, when LKB1 is absent. The major cell lines’ LKB1 expression levels were also analyzed (Supplementary Figure S3e). Hence, our data support that PIKE-A blocks AMPK phosphorylation via shielding AMPK from its upstream kinases.

PIKE-A inhibits AMPK phosphorylation by LKB1 or CaMKKβ. (a) Overexpression of LKB1 (wild-type or kinase-dead K78M), STRADα, and MO25α in the presence or absence of PIKE-A in A549 cells. AMPKα and ACC phosphorylation status and validation of transfected constructs were analyzed by immunoblotting. (b) LKB1 +/+ and −/− MEFs were infected with shPIKE adenovirus and AMPKα and ACC phosphorylation status and endogenous PIKE-A were analyzed by immunoblotting. (c) LKB1 +/+ and −/− MEFs were transfected with GST-PIKE-A or GST PIKE—FF. AMPK and ACC phosphorylation status and validation of transfected constructs in the transfected cells were analyzed by immunoblotting. (d and e) Purified GST-PIKE-A or FF recombinant proteins were incubated with immunoprecipitated myc-tagged AMPKα kinase-dead (K45R) proteins, which were then incubated with LKB1 (d) or active CaMKKβ (e) in kinase reaction buffer for 20 min at 30 °C. AMPK phosphorylation status and validation of the expression of transfected constructs and LKB1 (d) or active CaMKKβ (e) were analyzed by immunoblotting
PIKE-A mediates cell proliferation via suppressing AMPK activity
To investigate the biological consequence of AMPK inhibition by PIKE-A, we monitored cell proliferation in AMPKα−/− MEF, which was transfected with wt or constitutive active mutant of AMPKα, followed by PIKE-A adenovirus (Ad-PIKE-A) infection. We found that PIKE-A obviously promoted cell proliferation in AMPKα wt-transfected AMPKα−/− MEF. However, there was no significant increase in AMPKα constitutive active mutant-transfected MEFs (Figure 6a). Furthermore, we measured cell proliferation in the presence of AMPK pharmacological activator AICAR. Overexpression of PIKE-A elevated cell proliferation in LN229 GBM cells compared with control and this effect was attenuated by AICAR. The degree of inhibition by AICAR is partially higher in Ad-PIKE-A infected groups than in control virus- (Ad-Control) infected groups (Figure 6b, upper). Immunoblotting demonstrated that PIKE-A substantially inhibited AICAR-triggered AMPK phosphorylation (Figure 6b, lower). We made a similar observation in TP366 GBM cells as well (Supplementary Figure S4a). Depletion of PIKE-A from LN229 cells, followed by treatment with inhibitor compound C to block AMPK, showed that shPIKE dramatically suppressed LN229 cells proliferation and this inhibition was rescued by Compound C. The degree of promotion by Compound C is higher in Ad-shPIKE-infected groups than in Ad-Control-infected groups (Figure 6c, upper). Elimination of PIKE-A augmented AMPK phosphorylation, which was partly repressed by compound C (Figure 6c, lower). We made the similar observation in TP366 cells (Supplementary Figure S4b). These findings suggest that PIKE-A stimulates cell proliferation via inhibiting AMPK signaling. Previous study shows that glucose availability regulates cell-cycle checkpoint via AMPK/p53 signaling pathway.33 We wondered whether PIKE-A also mediates cell proliferation under the nutrient restriction. Accordingly, we found that both MEFs proliferated much faster in the presence of 25 mM glucose than 1 mM glucose and wild-type MEF grew much faster than PIKE−/− MEF (Supplementary Figure S4c). AICAR treatment prominently inhibited PIKE+/+ and PIKE−/− MEF cell proliferation (Supplementary Figure S4d). Therefore, our findings support that PIKE-A promotes cell proliferation, at least, partially through inhibiting the tumor suppressive action of AMPK pathway.

PIKE-A mediates cell proliferation via suppressing AMPK activity. (a) AMPKα −/− MEF was transfected with wt or constitutive active mutant of AMPKα, followed by PIKE-A adenovirus infection. Cell proliferation was determined by cell number of each cell line after 3 days seeding (n=3). The cell lysates were analyzed by immunoblotting with the indicated antibodies. The error bars represent mean values±S.D. from three replicates of each sample (**P<0.01). (b) Infection of LN229 cells with control and PIKE-A adenovirus, in the presence or absence of AICAR (0.2 mM). Cell proliferation was examined by cell number of each cell line after 3 days seeding (**P<0.01, n=3). The cell lysates were analyzed by immunoblotting with the indicated antibodies. (c) LN229 cells were infected with shRNA of PIKE-A adenovirus, followed by treatment with compound C (0.2 μM). Cell proliferation was examined by cell number of each cell line after 3 days seeding (**P<0.01, n=3). The cell lysates were analyzed by immunoblotting with the indicated antibodies
PIKE-A promotes tumorigenesis through inhibiting AMPK
To explore the biological consequence of PIKE-A inhibiting AMPK in tumorigenesis, we performed cell proliferation and colony formation assay by stably transfecting NIH3T3 with GFP vector, GFP-PIKE-A wt, and GFP-PIKE-A-FF mutant, respectively. In medium containing 0.5% serum, NIH3T3 with vector control barely proliferated. However, expression of PIKE-A wt conferred NIH-3T3 cells proliferation potential in low serum medium. As expected, PIKE-A strongly suppressed AMPK/ACC signaling cascades, consistent with the oncogenic functions of PIKE-A. In contrast, the PIKE-A FF mutant lost the ability to promote serum-independent growth (Figure 7a). We made the similar observations in PIKE-A wt or PIKE-A-FF stably transfected HeLa cells. Notably, PIKE-A-FF provoked Akt activation as strongly as PIKE-A wt, suggesting that PIKE-A predominantly exerts its oncogenic activity via suppressing AMPK pathway (Supplementary Figure S5a). AICAR evidently suppressed NIH3T3 stable cell proliferation, though PIKE-A wt stable cells grew faster than control and FF stable cells. On the other hand, Compound C elevated cell proliferation, and again, PIKE-A wt cell proliferated faster than both control and FF mutant cells (Supplementary Figures S5b and c). Colony formation assay demonstrated that PIKE-A wt induced anchorage-independent growth. By contrast, PIKE-A-FF mutant, which failed to bind or inhibit AMPK, was unable to induce anchorage-independent growth (Figure 7b). We made the similar observation in LKB1-deficient HeLa cells (Supplementary Figure S5d). These results indicate that PIKE-A may also exert the oncogenic transformation activity via inhibiting AMPK, which is regulated by Fyn-mediated tyrosine phosphorylation.

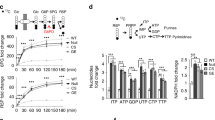
PIKE-A promotes tumorigenesis through inhibiting AMPK. (a) NIH3T3 cells were stably transfected with GFP, GFP-PIKE-A, and GFP-PIKE-A-FF. The cell proliferation was conducted in low (0.5%) serum. Cells were seeded at the same density and then their growth rates were determined and AMPK/ACC signalings were monitored. The error bars represent mean values±S.D. from three replicates of each sample (***P<0.001). (b) Colony formation assays were performed using NIH3T3 cells stably transfected with vector control or indicated constructs. Colonies were visualized with crystal violet staining and pictured (upper). Quantification of the colony number (lower). The error bars represent mean values±S.D. from three replicates of each sample (**P<0.01). (c) Immunohistochemistry staining of 15 human primary glioma with anti-p-AMPKα T172 and anti-PIKE-A. The summarized data are indicated in the right panel. (d) The proposed potential model for Fyn/PIKE-A signaling in suppressing AMPK pathway. Fyn inhibits AMPK signalings via phosphorylating both substrates LKB1 and PIKE-A. Fyn phosphorylation of PIKE-A stimulates its binding to AMPK, blocking AMPK phosphorylation and activation by the upstream kinase LKB1. Fyn phosphorylates LKB1 and sequestrates it from the substrate AMPK, inhibiting its activation
If PIKE-A provokes tumorigenesis via inhibiting the tumor suppressor AMPK’s activity, then one would expect a reduction of phospho-AMPK signal in human cancers with elevated PIKE-A expression levels. To test this possibility, we screened a panel of human primary gliomas with immunohistochemistry by staining with anti-PIKE-A or anti-p-AMPK antibody. We found that PIKE-A was strongly expressed (3+) in 10 and moderate (2+) in 5 GBMs. Its expression was predominantly localized in the cytoplasm. We also found that phospho-AMPK was weak or absent in nine cases and mild in six cases. Its expression was mostly noted in the cytoplasm with occasional cells showing nuclear expression. Of the 10 cases with strong PIKE-A, 8 had weak/absent and 2 had mild phospho-AMPK. Of the five cases with moderate PIKE-A, four had mild and one had weak/absent phospho-AMPK. Thus, in the GBM samples investigated, the expression of PIKE-A appeared to be inversely correlative to the AMPK phosphorylation (Figure 7c). Together, these results strongly support that PIKE-A binds AMPK and inhibits its tumor suppressive actions, which is regulated by Fyn-mediated phosphorylation of PIKE-A.
Discussion
In this report, we show that PIKE-A is an innovative binding partner of AMPK. PIKE-A directly associates with the kinase domain on AMPK α subunit via its GTPase domain. Interaction with PIKE-A shields AMPK from its upstream kinases, resulting in an inhibition of T172 phosphorylation and suppression of AMPK signaling. We provide several lines of evidence from pharmacological inhibition, Fyn knockout mouse tissues, siRNA of Fyn, and overexpression of Fyn-CA or KD constructs to demonstrate that Fyn phosphorylation of PIKE-A has an essential role in regulating its association with AMPK. Accordingly, blockade of PIKE-A phosphorylation by Fyn lessens its repression on AMPK signaling. Additionally, we show that PIKE-A promotes cancer cell proliferation and oncogenic transformation via inhibiting AMPK pathway. In alignment with these observations, we find that PIKE-A is highly expressed in most human glioblastomas and is tightly coupled to low levels of phospho-AMPK. Hence, our results support that PIKE-A negatively regulates AMPK signaling through direct interaction. Our previous studies demonstrate that Src also phosphorylates PIKE-A on its tyrosine residues, though Fyn reveals much stronger effect.30 Moreover, in cancer cell lines, c-Src signals through PKCa/PLC-γ/LKB1 to AMPK.36 Conceivably, besides Fyn, Src tyrosine kinase may crosstalk with AMPK via regulates PIKE-A phosphorylation.
AMPK controls cellular energy balance by promoting ATP-generating pathways such as fatty acid oxidation, while simultaneously inhibiting ATP-utilizing pathways, such as fatty acid synthesis and gluconeogenesis.10 AMPK achieves its regulatory functions either via direct and rapid phosphorylation of the metabolic enzymes or induces target gene expression. AMPK is negatively regulated by its binding partner CIDEA, which mediates its stability via ubiquitination and degradation. Accordingly, knockout of CIDEA in brown adipocyte tissue increases AMPK expression levels and activity.37 Consequently, CIDEA-deficient mice demonstrate lean phenotype and resistant to high fat diet (HFD)-induced obesity and diabetes.38 These phenotypes are almost identical to what we observed in PIKE-null mice. PIKE−/− mice exhibit a prominent phenotype of lipoatrophy and are resistant to HFD-induced obesity, liver steatosis, and diabetes. PIKE knockout mice also exhibit augmented lipid oxidation, which is accompanied by enhanced AMPK phosphorylation in both muscle and adipose tissues.28 Notably, these phenotypes are similar to that reported in Fyn-null mice, which also display a lean phenotype, upregulation of insulin sensitivity and activation of AMPK/ACC signaling.39 Thus, interaction between Fyn, PIKE-A, and AMPK forms a functional network to control both metabolism and cell proliferation.
Fyn directly phosphorylates LKB1 and triggers LKB1 nuclear translocation, leading to inhibition of AMPK.35 In the current study, we reveal a novel mechanism of Fyn-induced oncogenesis by promoting PIKE-A/AMPK interaction. We show that PIKE-A phosphorylation by Fyn is necessary for its interaction with AMPK (Figure 3). Moreover, we show that the AMPK inhibitory effect of PIKE-A is also mediated by Fyn phosphorylation. Abolishing PIKE-A phosphorylation by Fyn via point mutagenesis or depletion of Fyn is sufficient to alleviate its blockade of AMPK activation (Figure 4). Conceivably, Fyn exerts its oncogenic effects and inhibits AMPK activation via both LKB1 and PIKE-A pathways (Figure 5). In LKB1-deficeint cells (A549, HeLa, or LKB1−/− MEF), overexpression of PIKE-A wt alone strongly suppresses AMPK activation. However, the inhibitory activity is lost when its Fyn-phosphorylated sites are mutated (Figure 4), further suggesting that Fyn phosphorylation is indispensable for PIKE-A to block AMPK signaling pathway. Since both LKB1 and PIKE-A are substrates of Fyn, Fyn remains capable of antagonizing AMPK signaling cascades even if one of the pathways is crippled (LKB1−/− or PIKE−/− MEF). We show that PIKE-A inhibits AMPK phosphorylation by LKB1 through directly binding to the catalytic subunit of AMPK and shields it from the upstream kinases (Figure 5). Since LKB1 is constitutively active, depletion of PIKE-A relieves its blockade of AMPK, elicits AMPK T172 phosphorylation, and activates AMPK signaling (Figures 2 and 4). Therefore, in PIKE−/− MEF, depletion of Fyn or overexpression of kinase-dead Fyn-KD leads to a stronger AMPK phosphorylation than PIKE+/+ MEF (Figure 4).
PIKE-A acts as a proto-oncogene and is frequently amplified in numerous human cancers within the CDK4 amplicon.25 Indeed, from an automated network analysis on core pathways of glioma formation, PIKE-A has recently been confirmed as a driver gene of glioblastoma.40 Previous studies suggest that several signaling pathways may contribute to the oncogenic activity of PIKE-A. For instance, PIKE-A directly binds active Akt and stimulates its kinase activity to mediate cancer cell invasion and survival.24, 26 Further, PIKE-A activates nuclear factor-kappa B (NF-κB) and promotes prostate cancer progression.41 In the current report, we introduce an additional layer of molecular regulation on AMPK signaling pathway. We show that AMPK negatively mediates the oncogenic activity of PIKE-A in cell transformation and cell proliferation (Figures 6 and 7). Remarkably, we find an inverse correlation between PIKE-A expression levels and phospho-AMPK signals in human glioblastoma samples (Figure 7c). It is worthnoting that AMPK can be directly phosphorylated by insulin-activated Akt on α subunit of AMPK at both S485/S491 residues to inhibit ischemia-induced T172 phosphorylation.42 In addition, PIKE-A associates active Akt and escalates its kinase activity.26 Presumably, PIKE-A inhibits AMPK pathways through at least two different mechanisms: (1) direct and physical interaction with α subunit to block T172 phosphorylation by the upstream kinases and (2) the active Akt enhanced by PIKE-A phosphorylates the α subunit of AMPK to repress its T172 phosphorylation and subsequent activation. Nonetheless, the data from PIKE-FF mutant, which possesses Akt stimulatory effect but fails to bind or inhibit AMPK, demonstrate that PIKE-A exerts its oncogenic activity predominantly via repressing AMPK tumor suppressive pathway (Figure 7 and Supplementary Figure S5). Conceivably, PIKE-A promotes tumorigenesis through either elevating Akt activity, inhibiting AMPK signaling or both in some tumor types. Collectively, our findings support that AMPK is coordinately inhibited by PIKE-A, and these events are synchronized by the upstream tyrosine kinase Fyn, which phosphorylates both PIKE-A and tumor suppressor LKB1, resulting blockade of the tumor suppressive actions of AMPK.
Materials and Methods
Cell culture and reagents
HEK293, HeLa, LN229, U87MG, and TP366 cells were maintained in DMEM including 10% FBS. A549 cells were cultured in RPMI-1640 medium with 10% FBS. PIKE−/− MEF was isolated from an E13.5 embryo using trypsin digestion as previously described28 and were maintained in DMEM with 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin. Fyn +/+ and −/− MEF, LKB1 +/+ and LKB1−/− MEF, AMPKalpha WT and null MEF were maintained in DMEM with 10% FBS. All cells were maintained at 37 °C with 5% CO2 atmosphere in a humidified incubator.
AICAR, Metformin hydrochloride, and Calcium Ionophore A23187 were from Sigma-Aldrich (St. Louis, MO, USA) and control small-interfering RNA (siRNA) and siFyn were purchased from Dharmacon (Thermo Fisher Scientific Inc., Waltham, MA, USA). Control Adenovirus (Ad-Con), adenovirus overexpressing PIKE-A (Ad-PIKE-A), and adenovirus knocking down PIKE (Ad-shPIKE) and were prepared as previously reported.29 PP2 (Fyn inhibitor) and PP3 (Negative control for PP2) were purchased from Abcam (Cambridge, MA, USA). [γ-32P]ATP was from Perkin-Elmer (Waltham, MA, USA). Anti-phospho-AMPKα (Thr172), anti-AMPKα, anti-phospho-Acetyl-CoA Carboxylase (Ser79), anti-Acetyl-CoA Carboxylase, anti-phospho-p70 S6 Kinase (Thr389), anti-p70 S6 Kinase, p27 Kip1, anti-phospho-Akt (Ser473), anti-Akt, and anti-Fyn were from Cell Signaling Technology (CST, Danvers, MA, USA). Anti-p21, anti-phospho-Tyr antibody PY99, and PY20 were from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-β-actin, anti-GST-HRP, anti-GFP, anti-Flag, anti-HA, anti-myc, and MBP were from Sigma-Aldrich. SAMS peptide and Anti-Phosphoserine/threonine (p-S/T) antibody were from Abcam. Fyn knockout mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA), and PIKE knockout mice were developed in our laboratory as reported previously.28
Immunohistochmistry analysis of human primary gliomas
This study included 15 glioblastomas (GBMs) that were neurosurgically resected from 6 males and 9 females ranging in age from 41 to 76 years old, with an average age of 58.5 years. Nearly all GBMs were primary (de novo) tumors, with only one progressing from a preceding lower grade glioma (secondary GBM). Tumor tissues were formalin-fixed and paraffin-embedded, sectioned at 5 microns and stained by immunohistochemistry for PIKE-A and p-AMPK. Tissue sections were reviewed for intensity of staining as weak/absent (0), mild (1+), moderate (2+), or strong (3+).
Co-immunoprecipitation and in vitro binding assays
These methods were performed essentially as described previously.31
In vitro AMPK activity assay
After transfection, 500 μg protein from each sample was prepared and immunoprecipitated by adding 2 μl anti-GFP antibody and 25 μl of protein A-G agarose (Santa Cruz Biotechnology) at 4 °C for 3 h. After centrifugation (14 000 × g, 1 min), the beads were washed with lysis buffer and then twice with 10 × AMPK buffer (5 mM MOPS, pH 7.2, 2.5 mM β-glycerophosphate, 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2, 0.05 mM DTT). The AMPK activity was assayed by adding 50 μl of reaction mixtures, consisting of 5 μl of AMPK buffer, 10 μl of SAMS peptide (1 mg/ml), 10 μl of ATP working stock consisting of 0.1 μl of 100 mM ATP, 1 μl of [γ-32P] ATP, and 5 μl of 1 mM AMP and incubated at 30 °C for 15 min. The beads were quickly pelleted and 25 μl of supernatant was spotted onto P81 Whatman paper. The filter papers were then washed 4–5 times with 1% phosphoric acid. After the final wash, the filters were quickly dried and counted in a scintillation counter.
Cell proliferation and viability assay
For GBM cell viability assay, 2 × 104 cells were seeded in 6-well plate and incubated at 37 °C for indicated days. Cell numbers were counted by trypan blue exclusion under a microscope (× 40) at indicated times. For MTT cell viability assay of stable cells, 5 × 103 cells were seeded in 96-well plate 24 h before the assay starts and were cultured at 37 °C for 3 days. Cell viability was determined by using the MTT based Cell Growth Determination Kit (Sigma-Aldrich).
Colony formation assay
103 HeLa and NIH-3T3 stable cells were seeded onto 6-well plate and were maintained in DMEM supplemented with 10% fetal bovine serum for 2–3 weeks until foci were evident. Cells were fixed with 10% acetic acid and 10% methanol, and then colonies were stained with 1% crystal violet and counted. The photographs of colonies were taken with microscopes at × 5 magnification. The area and number of colonies were determined with Image-Pro 4.5 (Media Cybernetics, Inc., Bethesda, MD, USA) and Photoshop (Adobe Systems, Incorporated, San Jose, CA, USA) software.
Statistical analysis
Data shown are from one representative experiment of multiple independent experiments and are given as mean±S.D. Statistical analysis of significance (P-values) was based on two-tailed Student's t test.
Abbreviations
- ACC:
-
acetyl CoA carboxylase
- AICAR:
-
aminoimidazole-4-carboxymide-1-β-D-ribofuranoside
- AMPK:
-
AMP-activated protein kinase
- CA:
-
constitutively active
- CaMKKβ:
-
Ca2+/calmodulin-dependent kinase kinase β
- CIDEA:
-
cell-death inducing DFFA-like effector
- CNTF:
-
ciliary neurotrophic factor
- GBM:
-
glioblastoma
- GST:
-
glutathione S-transferase
- HFD:
-
high-fat diet
- KD:
-
kinase dead
- IL-6:
-
interleukin 6
- LKB1:
-
liver kinase B1
- MEF:
-
mouse embryonic fibroblast
- mTORC1:
-
mammalian target of rapamycin complex 1
- NFκB:
-
nuclear factor kappa B
- PIKE-A:
-
phosphoinositide 3-kinase enhancer A
- PKC:
-
protein kinase C
- PLC:
-
Phospholipase C
- STRAD:
-
Ste 20-related adaptor
- TNFα:
-
tumor necrosis factor α
- TZD:
-
thiozolidinedione
References
Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D . Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem 2007; 282: 32539–32548.
Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 1996; 271: 27879–27887.
Stein SC, Woods A, Jones NA, Davison MD, Carling D . The regulation of AMP-activated protein kinase by phosphorylation. Biochem J 2000; 345: 437–443.
Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 2004; 101: 3329–3335.
Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2003; 2: 28.
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003; 13: 2004–2008.
Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2005; 2: 9–19.
Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2005; 2: 21–33.
Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA . The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 2005; 280: 29060–29066.
Fogarty S, Hardie DG . Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta 2010; 1804: 581–591.
Dzamko NL, Steinberg GR . AMPK-dependent hormonal regulation of whole-body energy metabolism. Acta Physiol (Oxf) 2009; 196: 115–127.
Luo Z, Zang M, Guo W . AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol 2010; 6: 457–470.
Inoki K, Zhu T, Guan KL . TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115: 577–590.
Carretero J, Medina PP, Blanco R, Smit L, Tang M, Roncador G et al. Dysfunctional AMPK activity, signalling through mTOR and survival in response to energetic stress in LKB1-deficient lung cancer. Oncogene 2007; 26: 1616–1625.
Xiang X, Saha AK, Wen R, Ruderman NB, Luo Z . AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem Biophys Res Commun 2004; 321: 161–167.
Kim YH, Liang H, Liu X, Lee JS, Cho JY, Cheong JH et al. AMPKalpha modulation in cancer progression: multilayer integrative analysis of the whole transcriptome in Asian gastric cancer. Cancer Res 2012; 72: 2512–2521.
Hallstrom TC, Mori S, Nevins JR . An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell 2008; 13: 11–22.
Jansen M, Ten Klooster JP, Offerhaus GJ, Clevers H . LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev 2009; 89: 777–798.
Ollila S, Makela TP . The tumor suppressor kinase LKB1: lessons from mouse models. J Mol Cell Biol 2011; 3: 330–340.
Ye K, Hurt KJ, Wu FY, Fang M, Luo HR, Hong JJ et al. Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1N. Cell 2000; 103: 919–930.
Ye K, Snyder SH . PIKE GTPase: a novel mediator of phosphoinositide signaling. J Cell Sci 2004; 117: 155–161.
Hu Y, Liu Z, Ye K . Phosphoinositol lipids bind to phosphatidylinositol 3 (PI3)-kinase enhancer GTPase and mediate its stimulatory effect on PI3-kinase and Akt signalings. Proc Natl Acad Sci USA 2005; 102: 16853–16858.
Ye K . PIKE/nuclear PI 3-kinase signaling in preventing programmed cell death. J Cell Biochem 2005; 96: 463–472.
Ahn JY, Hu Y, Kroll TG, Allard P, Ye K . PIKE-A is amplified in human cancers and prevents apoptosis by up-regulating Akt. Proc Natl Acad Sci USA 2004; 101: 6993–6998.
Liu X, Hu Y, Hao C, Rempel SA, Ye K . PIKE-A is a proto-oncogene promoting cell growth, transformation and invasion. Oncogene 2007; 26: 4918–4927.
Ahn JY, Rong R, Kroll TG, Van Meir EG, Snyder SH, Ye K . PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J Biol Chem 2004; 279: 16441–16451.
Qi Q, He K, Liu X, Pham C, Meyerkord C, Fu H et al. Disrupting the PIKE-A/Akt interaction inhibits glioblastoma cell survival, migration, invasion and colony formation. Oncogene 2013; 32: 1030–1040.
Chan CB, Liu X, Jung DY, Jun JY, Luo HR, Kim JK et al. Deficiency of phosphoinositide 3-kinase enhancer protects mice from diet-induced obesity and insulin resistance. Diabetes 2010; 59: 883–893.
Chan CB, Liu X, He K, Qi Q, Jung DY, Kim JK et al. The association of phosphoinositide 3-kinase enhancer A with hepatic insulin receptor enhances its kinase activity. EMBO Rep 2011; 12: 847–854.
Tang X, Feng Y, Ye K . Src-family tyrosine kinase fyn phosphorylates phosphatidylinositol 3-kinase enhancer-activating Akt, preventing its apoptotic cleavage and promoting cell survival. Cell Death Differ 2007; 14: 368–377.
Tang X, Jang SW, Okada M, Chan CB, Feng Y, Liu Y et al. Netrin-1 mediates neuronal survival through PIKE-L interaction with the dependence receptor UNC5B. Nat Cell Biol 2008; 10: 698–706.
Tse MC, Liu X, Yang S, Ye K, Chan CB . Fyn regulates adipogenesis by promoting PIKE-A/STAT5a interaction. Mol Cell Biol 2013; 33: 1797–1808.
Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18: 283–293.
Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 2007; 9: 218–224.
Yamada E, Pessin JE, Kurland IJ, Schwartz GJ, Bastie CC . Fyn-dependent regulation of energy expenditure and body weight is mediated by tyrosine phosphorylation of LKB1. Cell Metab 2010; 11: 113–124.
Mizrachy-Schwartz S, Cohen N, Klein S, Kravchenko-Balasha N, Levitzki A . Up-regulation of AMP-activated protein kinase in cancer cell lines is mediated through c-Src activation. J Biol Chem 2011; 286: 15268–15277.
Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J et al. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J 2008; 27: 1537–1548.
Zhou Z, Yon Toh S, Chen Z, Guo K, Ng CP, Ponniah S et al. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat Genet 2003; 35: 49–56.
Bastie CC, Zong H, Xu J, Busa B, Judex S, Kurland IJ et al. Integrative metabolic regulation of peripheral tissue fatty acid oxidation by the SRC kinase family member Fyn. Cell Metab 2007; 5: 371–381.
Cerami E, Demir E, Schultz N, Taylor BS, Sander C . Automated network analysis identifies core pathways in glioblastoma. PLoS ONE 2010; 5: e8918.
Cai Y, Wang J, Li R, Ayala G, Ittmann M, Liu M . GGAP2/PIKE-a directly activates both the Akt and nuclear factor-kappaB pathways and promotes prostate cancer progression. Cancer Res 2009; 69: 819–827.
Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 2006; 281: 5335–5340.
Acknowledgements
We thank Dr. Paul Stein at SRI for Fyn+/+ and Fyn−/− MEFs and Dr. Viollet Benoit at Inserm for the AMPKα null MEFs. The work is supported by a grant from NIH (RO1 DK09092) to CBC and NIH (RO1 CA186918) to KY.
Author Contributions
SZ and KY contributed to conception and design; SZ, CBC, and KY contributed to development of methodology; SZ, WZ, CA, DJB, and KY contributed to acquisition of data (provided cells acquired and managed patients, provided facilities, etc.); SZ, CA, DJB, and KY contributed to analysis and interpretation of data (e.g., statistical analysis, biostatistics, and computational analysis); SZ, CBC, WZ, JC, HRL, and KY contributed to writing, review, and/or revision of the manuscript; SZ, WZ, CA, and DJB contributed to administrative, technical, or material support (i.e., reporting or organizing data, constructing databases); KY contributed to study supervision.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by X Lu
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article

Cite this article
Zhang, S., Qi, Q., Chan, C. et al. Fyn-phosphorylated PIKE-A binds and inhibits AMPK signaling, blocking its tumor suppressive activity. Cell Death Differ 23, 52–63 (2016). https://doi.org/10.1038/cdd.2015.66
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.66
This article is cited by
-
FYN: emerging biological roles and potential therapeutic targets in cancer
Journal of Translational Medicine (2023)
-
Advances in the expression and function of Fyn in different human tumors
Clinical and Translational Oncology (2023)
-
A novel chalcone derivative suppresses melanoma cell growth through targeting Fyn/Stat3 pathway
Cancer Cell International (2020)
-
FYN is required for ARHGEF16 to promote proliferation and migration in colon cancer cells
Cell Death & Disease (2020)
-
Cellular energy stress induces AMPK-mediated regulation of glioblastoma cell proliferation by PIKE-A phosphorylation
Cell Death & Disease (2019)
