
Abstract
Background:
Uterine leiomyomas from hereditary leiomyomatosis and renal cell cancer (HLRCC) patients are driven by fumarate hydratase (FH) inactivation or occasionally by mediator complex subunit 12 (MED12) mutations. The aim of this study was to analyse whether MED12 mutations and FH inactivation are mutually exclusive and to determine the contribution of MED12 mutations on HLRCC patients’ myomagenesis.
Methods:
MED12 exons 1 and 2 mutation screening and 2SC immunohistochemistry indicative for FH deficiency was performed on a comprehensive series of HLRCC patients’ (122 specimens) and sporadic (66 specimens) tumours. Gene expression analysis was performed using Affymetrix GeneChip Human Exon Arrays (Affymetrix, Santa Clara, CA, USA).
Results:
Nine tumours from HLRCC patients harboured a somatic MED12 mutation and were negative for 2SC immunohistochemistry. All remaining successfully analysed lesions (107/116) were deficient for FH. Of sporadic tumours, 35/64 were MED12 mutation positive and none displayed a FH defect. In global gene expression analysis FH-deficient tumours clustered together, whereas HLRCC patients’ MED12 mutation-positive tumours clustered together with sporadic MED12 mutation-positive tumours.
Conclusions:
Somatic MED12 mutations and biallelic FH inactivation are mutually exclusive in both HLRCC syndrome-associated and sporadic uterine leiomyomas. The great majority of HLRCC patients’ uterine leiomyomas are caused by FH inactivation, but incidental tumours driven by somatic MED12 mutations also occur. These MED12 mutation-positive tumours display similar expressional profiles with their sporadic counterparts and are clearly separate from FH-deficient tumours.
Similar content being viewed by others

Main
Uterine leiomyomas, or fibroids, are very common benign tumours among reproductive-aged women; nearly 70% are affected by these lesions before age 50 (Day Baird et al, 2003). Approximately one quarter of the patients suffer from symptoms such as abdominal pain, abnormal uterine bleeding, subfertility, and complications during pregnancy (Stewart, 2001). Uterine leiomyomas originate from the smooth muscle layer of the uterus and most are formed sporadically. Several independent studies on various populations have shown that ∼70% of uterine leiomyomas harbour specific mutations in mediator complex subunit 12 (MED12) (Mäkinen et al, 2011; Je et al, 2012; McGuire et al, 2012). All observed changes have been missense mutations or small in-frame insertions and deletions in exons 1 and 2 affecting a highly conserved area of the gene (Mäkinen et al, 2011; Je et al, 2012; McGuire et al, 2012; Kämpjärvi et al, 2014). Other recurrently, albeit less frequently, observed alterations include genomic rearrangements affecting HMGA2 in chromosome 12q15 and RAD51B in 14q23-24 (Ingraham et al, 1999; Sandberg, 2005). Recurrent non-random translocations and deletions have also been observed in other chromosomal areas, such as COL4A5 and COL4A6 locus in X chromosome, 6p21 (HMGA1), and 7q22 (Van de Ven, 1998; Sandberg, 2005; Mehine et al, 2013).
In addition to sporadic lesions, uterine leiomyomas are a feature of hereditary leiomyomatosis and renal cell cancer syndrome (HLRCC, OMIM #150800). HLRCC is a tumour predisposition syndrome caused by heterozygous germ-line mutations in fumarate hydratase (FH; Launonen et al, 2001; Tomlinson et al, 2002). The gene encodes fumarase enzyme, which catalyses the hydration of fumarate to L-malate in tricarboxylic acid cycle. FH is a tumour suppressor, and syndrome-associated lesions display biallelic FH inactivation through loss of heterozygosity (LOH) or inactivating mutation in the wild-type allele. In addition to uterine leiomyomas, FH mutations predispose individuals to cutaneous leiomyomas, and more rarely, to aggressive renal cell cancer.
Based on our recent preliminary results, somatic MED12 mutations and biallelic FH inactivation might exclude each other as drivers of leiomyomagenesis (Mäkinen et al, 2013; Mehine et al, 2013; Mäkinen et al, 2014). Here, our aim was to systematically analyse whether these phenomena are truly mutually exclusive and to clarify the role of MED12 in the tumourigenesis of HLRCC patients’ uterine leiomyomas. We collected a comprehensive series of HLRCC patients’ samples totalling 122 uterine leiomyomas from 27 individuals. Sixty-six sporadic conventional uterine leiomyomas were also included in the study. MED12 mutation status was analysed by direct sequencing and biallelic FH inactivation was determined by 2SC immunohistochemistry, which has proven to be an accurate and unambiguous method to detect FH deficiency (Bardella et al, 2011). The effects of MED12 and FH alterations on global expression profiles and clustering of the tumours were also studied.
Materials and methods
Subjects and ethical approval of the study
Twenty-seven Finnish HLRCC patients, representing 11 families, and 66 individuals with sporadic leiomyomas were included in the study. Altogether 188 uterine leiomyoma samples were collected, 122 tumours from HLRCC patients (89 formalin-fixed paraffin-embedded (FFPE) and 33 fresh frozen samples) and 66 tumours (FFPE samples) from individuals with sporadic uterine leiomyomas. Samples were collected either after signed informed consent or after the approval by the National Supervisory Authority for Welfare and Health. Sporadic samples were collected and anonymised after the approval by the director of the health care unit. The study was approved by the Ethics Review Board of the Hospital District of Helsinki and Uusimaa (HUS), Helsinki, Finland.
DNA/RNA extraction and sequencing
Genomic DNA was extracted from the FFPE samples with a standard phenol–chloroform extraction method or with NucleoSpin DNA FFPE XS Kit (Macherey-Nagel GmbH & Co KG, Düren, Germany), and from fresh frozen tissue samples with FastDNA Kit (MP Biomedicals LLC, Solon, OH, USA). Total RNA was extracted with TRI Reagent (Molecular Research Center Incorporated, Cincinnati, OH, USA). MED12 and FH mutation screenings were performed by direct sequencing. Sanger sequencing was carried out at the Institute of Molecular Medicine Finland, Technology Center, Helsinki, Finland, using an Applied Biosystems ABI3730 Automatic DNA Sequencer (Life technologies, Thermo Fisher Scientific, Waltham, MA, USA). Sequences were analysed both manually and with the Mutation Surveyor software (Softgenetics, State College, PA, USA). MED12 NM_005120.2 and FH NM_000143.3 were used as reference sequences. Primers used in the sequencing are listed in the Supplementary Table 1.
Loss of heterozygosity analysis
LOH at the FH locus was assessed from fresh frozen tumour samples harbouring both a germ-line FH mutation and a somatic MED12 mutation. LOH was analysed by sequencing five independent PCR reactions and comparing the heights of wild-type and mutant peaks in the chromatograms. LOH was scored when the height of wild-type allele peak was repeatedly lower than the mutant allele peak.
Tissue microarray construction
To determine the most suitable areas of the samples for tissue-microarray (TMA), haematoxylin and eosin staining was performed to sections cut from all FFPE samples. Tissue samples were analysed and representative tumour areas were marked by a gynaecological pathologist (RB). Four 0.8-mm tumour cores were punched from the original sample block with a manual tissue arrayer (MTA-I, Beecher Instruments, Sun Prairie, WI, USA) and inserted in TMA paraffin block. Normal myometrium samples were included in the TMA as internal controls.
Immunohistochemistry
Biallellic inactivation of FH was assessed with 2SC immunohistochemical staining performed with EnVision + kit (Dako, Carpinteria, CA, USA). Staining is based on the recognition of S-(2-succinyl) cysteine modified (succinated) proteins which are formed in FH deficient cells as a result of the accumulation of fumarate (Nagai et al, 2007; Bardella et al, 2011). Tissue sections (5 μm) were incubated with the anti-2SC antibody (1 : 2000) at +4 °C, o/n (FFPE samples) or at room temperature for an hour (fresh frozen samples). Antibody binding was detected with anti-rabbit horseradish peroxidase polymer. Stainings were assessed by a pathologist (RB) and the sample was scored as positive (+) when the great majority of cells displayed both nuclear and cytoplasmic staining indicating accumulation of fumarate and succinated proteins and thus biallelic inactivation of FH. Samples displaying no staining or rare samples showing low cytoplasmic positivity in single cells were scored as negative (−), indicating the cells to retain sufficient FH activity.
Gene expression profiling and hierarchical clustering analysis
Gene expression profiles of 14 leiomyomas from four HLRCC patients and three FH-deficient sporadic tumours and their corresponding normal myometrial tissue samples were analysed together with 58 uterine leiomyomas and corresponding normal samples from which we had both expression and whole-genome sequencing data in-house (19 MED12 mutation-positive and 39 MED12 and FH wild-type tumours) (Mehine et al, 2013, 2016). Gene expression data was constructed using Affymetrix GeneChip Human Exon 1.0 ST Arrays (Affymetrix) at the Biomedicum Functional Genomics Unit, Helsinki, Finland. The expression data was analysed with Partek Genomic Suite v. 6.5 (Partek Incorporated, St Louis, MO, USA) using re-mapped Brainarray Custom CDF files (HuEx10stv2_Hs_ENSG, Version 16). All samples were quantile-normalised by the Robust Multichip Average method and adjusted for probe sequence and GC-content. Unsupervised hierarchical clustering analysis (Cosine dissimilarity) was performed with 1% most variable genes (n=372), defined by the coefficient of variation calculated across all tumour samples.
Statistical analyses
R software, version 3.0.2 (R Foundation for Statistical Computing, Vienna, Austria, www.r-project.org), and Python, version 2.7 (Python Software Foundation, Wilmington, DE, USA, www.python.org), were utilised for statistical analyses. Differences in the frequencies of MED12 mutations and biallelic FH inactivation between uterine leiomyomas from HLRCC patients and sporadic conventional uterine leiomyomas were calculated using Fisher’s exact test (two-sided P-value). Mutual exclusiveness of MED12 mutations and FH deficiency was evaluated utilising a permutation test. A total of n=1 000 000 permutations were performed and mutations were randomly redistributed between samples in each permutation. Empirical P-value was computed as P=(1+k)/n, where k is the number of permutations where at least one sample presented both MED12 mutation and FH deficiency. The statistical significance of many MED12 mutation-positive tumours co-occurring in HLRCC patient My31 was evaluated similarly as above by randomly redistributing tumours to patients (n=10 000 000 permutations). Empirical P-value was then calculated as P=(1+k)/n, where k is the number of permutations where at least one patient with six or more MED12 mutation-positive tumours and no FH-deficient tumours was observed.
Results
Mutation screening
One hundred and twenty-two uterine leiomyomas from 27 HLRCC patients were screened for somatic mutations in exons 1 and 2 of MED12. Of these, 34 tumours had been previously screened for exon 2 mutations (Mäkinen et al, 2013). Both exons were successfully sequenced in 83 FFPE samples and in all 33 fresh frozen samples (116/122; 95%). The quality of DNA obtained from six FFPE blocks of patient D6 (dating back to year 1979) was not sufficient for sequencing, and these samples were excluded from mutation screening.
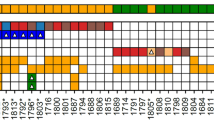
Altogether nine tumours from four different HLRCC patients had MED12 mutations (9/116; 7.8%) (Supplementary Table 2). Patients B3, B7, and E1 had one MED12 mutation-positive tumour each, while the majority of their tumours were MED12 wild type (1/3, 1/6, and 1/6, respectively) (Table 1). Surprisingly, all six uterine leiomyomas from patient My31 harboured different MED12 mutation. Coding exons of FH were sequenced from all these six tumours, but no second hit was observed (Table 1 and Figure 1). Occurrence of this many sporadic MED12 mutation-positive lesions in HLRCC patient is highly unlikely (P=2 × 10−7, permutation test with 10 000 000 permutations).

FH and MED12 mutation status of six uterine leiomyomas form HLRCC patient My31. All six fresh frozen tumour samples retain heterozygosity at the FH locus and display different somatic MED12 mutation.
Sixty-four sporadic conventional uterine leiomyomas (64/66; 97%) were successfully sequenced for MED12 exons 1 and 2 (45 reported previously in (Mäkinen et al, 2013). Of these, 35/64 (55%) harboured a somatic MED12 mutation (Supplementary Table 3).
Immunohistochemistry
All 122 HLRCC-associated uterine leiomyomas and 66 sporadic tumours were successfully analysed for FH deficiency utilising 2SC immunohistochemistry. Positive staining indicating biallelic FH inactivation was detected in 113 uterine leiomyomas from HLRCC patients (113/122; 92.6%, including six tumours from patient D6 excluded from MED12 mutation screening). Nine uterine leiomyomas (one tumour from patients B3, B7, and E1 each, and all six tumours from My31) displayed negative 2SC immunostaining implicating the other FH allele to be intact and tumours to be proficient for FH activity (9/122; 7.4%; Figure 2 and Supplementary Table 2). These same tumours were observed to harbour somatic MED12 mutations in the mutation screening (Table 1). All 66 sporadic tumours showed negative 2SC staining (Supplementary Table 3).

Haematoxylin-eosin staining and 2SC immunohistochemical staining of four uterine leiomyomas from two HLRCC patients (original magnification, × 200). Tumours deficient for FH and without a MED12 mutation (top panel; patient B3 tumour m3 and patient E1 tumour m7) display clear positive 2SC staining, whereas tumours harbouring a MED12 mutation and not displaying biallelic FH inactivation (bottom panel; patient B3 tumour m2 and patient E1 tumour m10) are negative.
Based on mutation screening and results from 2SC immunohistochemistry, the difference in MED12 mutation and biallelic FH inactivation frequencies between uterine leiomyomas from HLRCC patients and sporadic conventional uterine leiomyomas is highly significant (P<2.2 × 10−16). Data also shows these events to be mutually exclusive (P=1 × 10-6, permutation test with 1 000 000 permutations).
Gene expression profiling
Gene expression profiling of 26 MED12 mutation-positive, 10 FH-deficient, and 39 MED12 and FH wild-type tumours and their corresponding normal myometrium tissue samples revealed that the majority of leiomyomas clustered according to the mutation status of FH and MED12 (Figure 3). Leiomyomas harbouring both a germ-line FH mutation and a somatic MED12 mutation displayed expression signatures similar to those with only a MED12 mutation, instead of clustering with the FH-deficient leiomyomas. All myometrium tissue samples clustered together regardless of their germ-line mutation status.

Unsupervised hierarchical clustering of 75 uterine leiomyomas and their corresponding normal myometrium tissue samples. Analysis included 26 MED12 mutation-positive (green), 10 FH-deficient (red), 39 MED12 and FH wild-type tumours, (grey), and 48 corresponding normal myometrium tissue samples (brown). Samples from HLRCC patients, harbouring a germ-line FH mutation, are marked with black. MED12 mutation-positive and FH-deficient tumours clustered according to their mutation status. Leiomyomas with both a germ-line FH mutation and a somatic MED12 mutation (My31 m1-m6 and B7 m1; red boxes) clustered with sporadic MED12 mutation-positive leiomyomas. Germ-line FH mutation did not affect the clustering of normal myometrium samples, all of which clustered together.
Discussion
Most uterine leiomyomas develop sporadically and in the majority (∼70%) tumourigenesis is driven by very specific mutations in exons 1 or 2 of MED12. Simple chromosomal changes and chromothripsis-like, more complex chromosomal rearrangements affecting specific loci such as HMGA2 (chromosome 12) or COL4A5 and COL4A6 (chromosome X), have also been suggested as driver aberrations in these tumours (Van de Ven, 1998; Sandberg, 2005; Mehine et al, 2013). Thus far, none of these alterations have been reported to occur simultaneously in the same lesion (Markowski et al, 2012; Mehine et al, 2013; Bertsch et al, 2014; Mehine et al, 2016).
HLRCC syndrome, caused by heterozygous germ-line mutations in FH, is one of the few known hereditary conditions that predispose women to uterine leiomyomas. Loss of the wild-type allele leads to FH inactivation and subsequent tumourigenesis. To determine the frequency of MED12 mutation-positive tumours among HLRCC patients, and to analyse whether somatic MED12 mutations and biallelic FH inactivation are mutually exclusive, we performed MED12 exons 1 and 2 mutation screening and 2SC immunohistochemical staining indicative for FH deficiency, in both HLRCC-associated and sporadic uterine leiomyomas.
In a comprehensive series of 122 HLRCC-associated uterine leiomyomas, 116 tumours were successfully analysed with both methods. The great majority of tumours (107/116; 92%) were wild type for MED12 and showed 2SC positivity indicating them as syndrome-associated FH-deficient lesions. Twenty-three out of 27 HLRCC patients (23/27; 85%) had only FH-deficient uterine leiomyomas, with number of tumours ranging from 1 to 15 per patient. Nine uterine leiomyomas (9/116; 7.8%) from four different HLRCC patients had somatic MED12 mutations. All these tumours displayed negative 2SC immunohistochemical staining indicating FH proficiency; these tumours are most likely sporadic. Three patients had only one MED12 mutation-positive uterine leiomyoma in addition to several syndrome-associated tumours, while all six tumours of patient My31 were MED12 mutation positive. Overall, these results confirm previous observations that most uterine leiomyomas in HLRCC patients are syndrome-associated and result from biallelic FH inactivation. Incidental sporadic tumours do, however, occur in HLRCC patients, yet these lesions account for only a small proportion of tumours. HLRCC patients tend to develop uterine leiomyomas at younger age than women with sporadic tumours (Lehtonen, 2011). Syndrome-associated tumours are also suggested to be more numerous and symptomatic, leading to hysterectomies at an earlier age, among women with HLRCC (Lehtonen, 2011). This might, to some extent, explain the rarity of sporadic lesions, which are known to reach the incidence peak towards the end of women’s reproductive age. Another possible explanation is purely stochastic: as the remaining FH allele can be inactivated through several distinct mechanisms (missense or nonsense mutation, small insertion, or deletion leading to a shift in the reading frame, larger chromosomal aberration), it may be that one of these events happens more likely than a single, highly specific MED12 mutation.
Altogether 66 sporadic conventional uterine leiomyomas were included in the study. Sixty-four were successfully analysed for MED12 mutations and the majority (35/64; 55%) were mutation positive. This is in agreement with previous studies, where mutation frequencies have varied between 48 and 92% (Mehine et al, 2014). Also the mutation spectrum with 26 highly specific missense mutations and 9 small in-frame deletions resembles previous observations. All sporadic tumours showed negative 2SC immunostaining and were thus FH proficient. This does not exclude the possibility of an underlying heterozygous germ-line FH mutation or the presence of one somatic alteration in some of the samples, but it shows that biallelic FH inactivation is a rare molecular driver event in sporadic uterine leiomyomas. This is in line with previous analyses, which have shown that the contribution of FH alterations on sporadic uterine leiomyomas and sporadic counterparts of other HLRCC-associated tumours is in a minor role (Kiuru et al, 2002; Lehtonen et al, 2004; Mehine et al, 2013; Harrison et al, 2015). Taken together, our results on an extensive HLRCC patients’ sample series and sporadic tumours confirm the previous observations that biallelic FH inactivation and MED12 mutations are mutually exclusive.
When sporadic MED12 mutation-positive tumours were observed in HLRCC patients, these were typically individual lesions among several FH-deficient tumours. Interestingly, all six tumours from one HLRCC patient, My31, showed FH proficiency and instead of losing the wild-type FH allele, each tumour harboured a MED12 mutation. All tumours displayed different MED12 mutations thus excluding the possibility of tumour dissemination. These six tumours had been obtained at hysterectomy, which was performed when the patient was 49. She had been under surveillance due to uterine leiomyomas since the age of 28, and several lesions had been removed in two myomectomies at ages 36 and 40. We were able to collect 24 FFPE uterine leiomyoma specimens from these operations, and we analysed them with direct sequencing and 2SC immunohistochemistry. Twenty-three out of 24 specimens (96%) were considered to represent sporadic tumours due to both MED12 mutation and negative 2SC staining. Only one tumour was wild type for MED12 and showed positive 2SC staining, and was thus considered syndrome-associated. The germ-line FH mutation in patient My31 is a nonsense mutation affecting the last exon of the gene (c.1439C>G; Ser480X). The same mutation has been previously observed in another Finnish HLRCC patient with both uterine and cutaneous leiomyomas (unpublished data). This patient had a hysterectomy due to multiple leiomyomas in 1968 at the age of 31. Uterine leiomyoma samples were no longer available, but we were able to collect nine cutaneous leiomyoma samples from the pathology department’s archives. All these tumours were FH deficient and thus considered syndrome-associated. In addition, other syndrome-associated nonsense and frameshift mutations have been reported in the last exon of FH, including one affecting Ser480 and others affecting nearby amino acids Lys477, Gly490, and Trp500 (Bayley et al, 2008). Intriguing and highly unlikely (P=2 × 10−7) observation of several sporadic MED12 mutation-positive lesions in HLRCC patient My31 implicates the presence of an additional genetic factor that either protects the patient from the loss of the FH wild-type allele or predisposes her to somatic MED12 mutations.
Unsupervised hierarchical clustering analysis of gene expression data revealed that FH-deficient and MED12 mutation-positive leiomyomas display distinct expression profiles. This is not surprising considering the severe metabolic stress FH inactivation exposes the cells to, and similar results have been reported also previously (Vanharanta et al, 2006; Mehine et al, 2013, 2016). We included both HLRCC syndrome-associated and sporadic FH-deficient tumours in the analysis, and all of these tumours clustered together irrespective of whether the inactivation had occurred through one germ line and one somatic or through two somatic mutational events. Interestingly, all six tumours of patient My31, which harbour both one germ-line FH mutation and a somatic MED12 mutation, clustered together with MED12 mutation-positive tumours and displayed expression signatures typically seen in sporadic MED12 mutation-positive leiomyomas. Also all normal tissue samples clustered together irrespective of germ-line FH mutation status. These results confirm that one active FH allele is sufficient to maintain the metabolic balance within the cell.
To conclude, HLRCC patients’ uterine leiomyomas can be formed through at least two distinct molecular mechanisms, biallelic FH inactivation and somatic MED12 mutations. The great majority of HLRCC patients’ tumours use the former pathway, while MED12 mutations are the most frequent alterations in sporadic tumours. Distinct molecular mechanisms within HLRCC patients’ uterine leiomyomas and sporadic tumours may affect treatment options of these patients in the future. Our results also confirm that biallelic FH inactivation and MED12 mutations are mutually exclusive both within HLRCC syndrome associated and sporadic uterine leiomyomas.
Change history
14 June 2016
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bardella C, El-Bahrawy M, Frizzell N, Adam J, Ternette N, Hatipoglu E, Howarth K, O'Flaherty L, Roberts I, Turner G, Taylor J, Giaslakiotis K, Macaulay VM, Harris AL, Chandra A, Lehtonen HJ, Launonen V, Aaltonen LA, Pugh CW, Mihai R, Trudgian D, Kessler B, Baynes JW, Ratcliffe PJ, Tomlinson IP, Pollard PJ (2011) Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol 225: 4–11.
Bayley JP, Launonen V, Tomlinson IP (2008) The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet 9: 20.
Bertsch E, Qiang W, Zhang Q, Espona-Fiedler M, Druschitz S, Liu Y, Mittal K, Kong B, Kurita T, Wei JJ (2014) MED12 and HMGA2 mutations: two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod Pathol 27: 1144–1153.
Day Baird D, Dunson DB, Hill MC, Cousins D, Schectman JM (2003) High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol 188: 100–107.
Harrison WJ, Andrici J, Maclean F, Madadi-Ghahan R, Farzin M, Sioson L, Toon CW, Clarkson A, Watson N, Pickett J, Field M, Crook A, Tucker K, Goodwin A, Anderson L, Srinivasan B, Grossmann P, Martinek P, Ondic O, Hes O, Trpkov K, Clifton-Bligh RJ, Dwight T, Gill AJ (2015) Fumarate hydratase-deficient uterine leiomyomas occur in both the syndromic and sporadic settings. Am J Surg Pathol 40 (5): 599–607.
Ingraham SE, Lynch RA, Kathiresan S, Buckler AJ, Menon AG (1999) hREC2, a RAD51-like gene, is disrupted by t(12;14) (q15;q24.1) in a uterine leiomyoma. Cancer Genet Cytogenet 115: 56–61.
Je EM, Kim MR, Min KO, Yoo NJ, Lee SH (2012) Mutational analysis of MED12 exon 2 in uterine leiomyoma and other common tumors. Int J Cancer 131: E1044–E1047.
Kämpjärvi K, Park MJ, Mehine M, Kim NH, Clark AD, Bützow R, Böhling T, Böhm J, Mecklin JP, Järvinen H, Tomlinson IP, van der Spuy ZM, Sjöberg J, Boyer TG, Vahteristo P (2014) Mutations in exon 1 highlight the role of MED12 in uterine leiomyomas. Hum Mutat 35: 1136–1141.
Kiuru M, Lehtonen R, Arola J, Salovaara R, Järvinen H, Aittomäki K, Sjöberg J, Visakorpi T, Knuutila S, Isola J, Delahunt B, Herva R, Launonen V, Karhu A, Aaltonen LA (2002) Few FH mutations in sporadic counterparts of tumor types observed in hereditary leiomyomatosis and renal cell cancer families. Cancer Res 62: 4554–4557.
Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, Sistonen P, Herva R, Aaltonen LA (2001) Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 98: 3387–3392.
Lehtonen HJ (2011) Hereditary leiomyomatosis and renal cell cancer: update on clinical and molecular characteristics. Fam Cancer 10: 397–411.
Lehtonen R, Kiuru M, Vanharanta S, Sjöberg J, Aaltonen LM, Aittomäki K, Arola J, Bützow R, Eng C, Husgafvel-Pursiainen K, Isola J, Järvinen H, Koivisto P, Mecklin JP, Peltomäki P, Salovaara R, Wasenius VM, Karhu A, Launonen V, Nupponen NN, Aaltonen LA (2004) Biallelic inactivation of fumarate hydratase (FH) occurs in nonsyndromic uterine leiomyomas but is rare in other tumors. Am J Pathol 164: 17–22.
Mäkinen N, Mehine M, Tolvanen J, Kaasinen E, Li Y, Lehtonen HJ, Gentile M, Yan J, Enge M, Taipale M, Aavikko M, Katainen R, Virolainen E, Böhling T, Koski TA, Launonen V, Sjöberg J, Taipale J, Vahteristo P, Aaltonen LA (2011) MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science 334: 252–255.
Mäkinen N, Vahteristo P, Bützow R, Sjöberg J, Aaltonen LA (2014) Exomic landscape of MED12 mutation-negative and -positive uterine leiomyomas. Int J Cancer 134: 1008–1012.
Mäkinen N, Vahteristo P, Kämpjärvi K, Arola J, Bützow R, Aaltonen LA (2013) MED12 exon 2 mutations in histopathological uterine leiomyoma variants. Eur J Hum Genet 21 (11): 1300–1303.
Markowski DN, Bartnitzke S, Loning T, Drieschner N, Helmke BM, Bullerdiek J (2012) MED12 mutations in uterine fibroids-their relationship to cytogenetic subgroups. Int J Cancer 131 (7): 1528–1536.
McGuire MM, Yatsenko A, Hoffner L, Jones M, Surti U, Rajkovic A (2012) Whole exome sequencing in a random sample of north american women with leiomyomas identifies MED12 mutations in majority of uterine leiomyomas. PLoS One 7: e33251.
Mehine M, Kaasinen E, Heinonen H, Mäkinen N, Kämpjärvi K, Sarvilinna N, Aavikko M, Vähärautio A, Pasanen A, Bützow R, Heikinheimo O, Sjöberg J, Pitkänen E, Vahteristo P, Aaltonen LA (2016) Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proc Natl Acad Sci U S A 113: 1315–1320.
Mehine M, Kaasinen E, Mäkinen N, Katainen R, Kämpjärvi K, Pitkänen E, Heinonen HR, Bützow R, Kilpivaara O, Kuosmanen A, Ristolainen H, Gentile M, Sjöberg J, Vahteristo P, Aaltonen LA (2013) Characterization of uterine leiomyomas by whole-genome sequencing. N Engl J Med 369: 43–53.
Mehine M, Mäkinen N, Heinonen HR, Aaltonen LA, Vahteristo P (2014) Genomics of uterine leiomyomas: insights from high-throughput sequencing. Fertil Steril 102: 621–629.
Nagai R, Brock JW, Blatnik M, Baatz JE, Bethard J, Walla MD, Thorpe SR, Baynes JW, Frizzell N (2007) Succination of protein thiols during adipocyte maturation: a biomarker of mitochondrial stress. J Biol Chem 282: 34219–34228.
Sandberg AA (2005) Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: leiomyoma. Cancer Genet Cytogenet 158: 1–26.
Stewart EA (2001) Uterine fibroids. Lancet 357: 293–298.
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomäki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA Multiple Leiomyoma Consortium (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30: 406–410.
Van de Ven WJ (1998) Genetic basis of uterine leiomyoma: involvement of high mobility group protein genes. Eur J Obstet Gynecol Reprod Biol 81: 289–293.
Vanharanta S, Pollard PJ, Lehtonen HJ, Laiho P, Sjöberg J, Leminen A, Aittomäki K, Arola J, Kruhoffer M, Orntoft TF, Tomlinson IP, Kiuru M, Arango D, Aaltonen LA (2006) Distinct expression profile in fumarate-hydratase-deficient uterine fibroids. Hum Mol Genet 15: 97–103.
Acknowledgements
We thank Sini Nieminen, Inga-Lill Svedberg, Iina Vuoristo, Alison Ollikainen, and Elina Pörsti for their excellent technical assistance. This study was supported by the Academy of Finland (Finnish Center of Excellence in Cancer Genetics Research, grant number 250345 for LAA, and Academy Research Fellow grants 260370 and 292769 for PV), the Sigrid Jusélius Foundation, the Cancer Society of Finland, and the Biomedicum Helsinki Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kämpjärvi, K., Mäkinen, N., Mehine, M. et al. MED12 mutations and FH inactivation are mutually exclusive in uterine leiomyomas. Br J Cancer 114, 1405–1411 (2016). https://doi.org/10.1038/bjc.2016.130
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.130
Keywords
This article is cited by
-
The Mediator Complex Subunit 12 (MED-12) Gene and Uterine Fibroids: a Systematic Review
Reproductive Sciences (2024)
-
Uterine leiomyoma with RAD51B::NUDT3 fusion: a report of 2 cases
Virchows Archiv (2023)
-
CYP24A1 expression analysis in uterine leiomyoma regarding MED12 mutation profile
Archives of Gynecology and Obstetrics (2021)
-
Characterization of MED12, HMGA2, and FH alterations reveals molecular variability in uterine smooth muscle tumors
Molecular Cancer (2017)
-
Global metabolomic profiling of uterine leiomyomas
British Journal of Cancer (2017)
