
Abstract
Aim:
Glyoxalase I (GLOI), a glutathione (GSH)-dependent enzyme, is overexpressed in tumor cells and related to multi-drug resistance in chemotherapy, making GLOI inhibitors as potential anti-tumor agents. But the most studied GSH analogs exhibit poor pharmacokinetic properties. The aim of this study was to discover novel non-GSH analog GLOI inhibitors and analyze their binding mechanisms.
Methods:
Mouse GLOI (mGLOI) was expressed in BL21 (DE3) pLysS after induction with isopropyl-β-D-1-thiogalactopyranoside and purified using AKTA FPLC system. An in vitro mGLOI enzyme assay was used to screen a small pool of compounds containing carboxyl groups. Crystal structure of the mGLOI-inhibitor complex was determined at 2.3 Å resolution. Molecular docking study was performed using Discovery Studio 2.5 software package.
Results:
A natural compound 18-β-glycyrrhetinic acid (GA) and its derivative carbenoxolone were identified as potent competitive non-GSH analog mGLOI inhibitors with Ki values of 0.29 μmol/L and 0.93 μmol/L, respectively. Four pentacyclic triterpenes (ursolic acid, oleanolic acid, betulic acid and tripterine) showed weak activities (mGLOI inhibition ratio <25% at 10 μmol/L) and other three (maslinic acid, corosolic acid and madecassic acid) were inactive. The crystal structure of the mGLOI-GA complex showed that the carboxyl group of GA mimicked the γ-glutamyl residue of GSH by hydrogen bonding to the glutamyl sites (residues Arg38B, Asn104B and Arg123A) in the GSH binding site of mGLOI. The extensive van der Waals interactions between GA and the surrounding residues also contributed greatly to the binding of GA and mGLOI.
Conclusion:
This work demonstrates a carboxyl group to be an important functional feature of non-GSH analog GLOI inhibitors.
Similar content being viewed by others

Introduction
Glyoxalase I (GLOI, EC 4.4.1.5) is the first and the rate-limiting glutathione (GSH)-dependent enzyme in the glyoxalase system, which functions as a ubiquitous detoxification pathway by catalyzing the conversion of toxic α-oxoaldehydes to nontoxic D-lactate1. Structurally, mammalian GLOI is a zinc-containing metalloenzyme with two active sites located at the homodimer interface2. Each active site of GLOI is composed of a GSH binding site (the glycyl site is characterized by Lys150A, Gly155A, Lys156A, Leu160A and Phe162A and the glutamyl site is characterized by Arg37B, Asn103B and Arg122A), a Zn2+ catalytic site (Zn2+ and the Zn2+-coordinating residues Gln32, His126 and Glu172) and a hydrophobic site (Leu69B, Leu92B, Met157A, Met179A, Met183A and Phe71B)3,4.
Abnormal overexpression or higher activity of GLOI has been observed in many tumors and is related to multi-drug resistance (MDR) in cancer chemotherapy5. Inhibition of GLOI resulted in the accumulation of α-oxoaldehydes at cytotoxic levels2 and reverse MDR, and thus GLOI inhibitors have been proposed as efficient anti-tumor agents and have been recognized for their anti-protozoal and anti-bacterial purposes for many years6,7,8,9. The traditional and most studied GLOI inhibitors are analogs of the coenzyme GSH, which exhibits efficient inhibition in vitro3. However, these GSH analog inhibitors suffer from poor pharmacokinetic properties and are difficult to use as a lead structure for the further design of small molecule GLOI inhibitors due to their structural similarity to GSH10,11. Moreover, these highly potent GSH analog inhibitors are not able to induce efficient cytotoxic concentrations of α-oxoaldehydes in tumor cells because of their degradation by other GSH-dependent enzymes12 and metabolism by aldose reductase in vivo13,14. Thus, a search of new types of scaffolds for GLOI inhibition with better pharmacokinetic properties is necessary.
At present, non-GSH analog inhibitors, such as methyl-gerfelin (MGI)15, azaindole-substituted N-hydroxypyridones16, flavonoids10, curcumin and its derivatives17, and nonsteroidal anti-inflammatory drugs (NSAIDs)5, have been identified or synthesized (see Supplementary Table S1 for representative structures and inhibition levels of GLOI inhibitors). Crystallographic studies indicate that the ability to chelate Zn2+ at the active center of GLOI plays an essential role for the high inhibitory activity of GSH analog inhibitors (GIP)3 and non-GSH analog inhibitors (MGI15 and azaindole-substituted N-hydroxypyridones16). Docking and molecular dynamic simulation analyses also suggest that coordination with Zn2+ is required for the inhibition of GLOI by curcumin, NSAIDs and flavonoids10,18,19. However, metal chelation is generally undesirable in drug development because of the high potential for off-target effects in the human body20,21.
In our recent work, we determined the crystal structure of mouse glyoxalase I (mGLOI) complexed with indomethacin (inhibition constant (Ki)=18 μmol/L, PDB ID: 4KYK) and zopolrestat (Ki=1.2 μmol/L, PDB ID: 4KYH), revealing a novel inhibitor-GLOI binding mode22. In these two complex structures, the carboxyl groups of indomethacin and zopolrestat mimic the glycyl and γ-glutamyl residue moieties of GSH to form hydrogen bonds with the glycyl and glutamyl sites, respectively, in the GSH binding site22. In this study, we propose the existence of a novel class of non-GSH analog GLOI inhibitors with a carboxyl group as an important functional feature. To confirm our proposal, we screened a small pool of compounds containing a carboxyl group using an in vitro assay. Furthermore, the binding mode and molecular mechanism of potential GLOI inhibitors were explored using X-ray crystallographic analysis.
Materials and methods
Protein expression, purification and in vitro enzyme assay
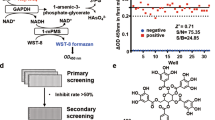
The mGLOI plasmid was the kindly gift of Dr Hideo OKUMURA, Advanced Science Institute, RIKEN, Japan. The mGLOI was expressed in BL21 (DE3) pLysS at 25 °C after induction with 0.1 mmol/L isopropyl-β-D-1-thiogalactopyranoside and purified on a HiTrap Q column using an AKTA FPLC system. The protein concentration was determined using a NanoDrop 2000c spectrophotometer.
The activity of recombinant mGLOI was determined by monitoring the changes in absorbance at 240 nm at 30 °C with a thermostated spectrophotometer, as described previously18,23. The test compound was preincubated with 0.10 mol/L potassium phosphate (pH 7.0) containing hemithioacetal (MG-SG) for 20 min at 30 °C. Different concentrations of methylglyoxal and GSH were calculated and varied by using the equation constant Kd=3.0 mmol/L to obtain the desired concentration of MG-SG. Excess free GSH in the assay was kept at 0.10 mmol/L. The reaction was initiated by the addition of mGLOI (3.0 nmol/L) to the reaction system. The Ki values of inhibitors were evaluated using a Dixon plot showing the reciprocal of enzyme velocity against the inhibitor concentration at various concentrations of the substrate.
Protein crystallization, data collection and structure deter-mination
The apo mGLOI crystals were obtained at 289 K in hanging droplets consisting of 2 μL of protein solution (20 mg/mL) mixed with 2 μL of reservoir solution consisting of 50 mmol/L 2-(N-morpholino)ethanesulfonic acid (MES; pH 5.8), 30%-32% PEG2000 and 0.10 mol/L or 0.15 mol/L NaCl. Apo mGLOI crystals were soaked in a saturated solution of GA containing 32.5% N,N-dimethylformamide and the corresponding reservoir solution. X-ray data were collected at 100 K on an in-house Oxford Diffraction Xcalibur Nova diffractometer. The structure was determined with CCP424, Phenix25 and COOT26 (PDB ID: 4PV5; for data collection and refinement statistics, see Supplementary Table S2).
Molecular docking experiment
All docking experiments were performed using the Discovery Studio 2.5 software package (Accelrys Software Inc, San Diego, CA, USA). The protocol was generated using the CDOCKER protocol without the crystal waters. Before docking, hydrogen atoms were added to the unoccupied valence of the heavy atoms of the protein. The molecules used for docking were downloaded from the PubChem Compound database (http://www.ncbi.nlm.nih.gov/pccompound/). Prior to docking, the protocol was demonstrated to be reliable by redocking the inhibitor of each PDB file to its own receptor (PDB IDs: 1QIN, 2ZA0, and 4KYH) in terms of global conformation, orientation and root mean square deviation value (<1.5 Å).
Results
Discovery of GA as a potent in vitro GLOI inhibitor
To search for GLOI inhibitors with a carboxyl group feature, we screened a small pool of compounds containing carboxyl groups using an in vitro assay. Among this pool, GA was found to be a potent competitive mGLOI inhibitor with a Ki of 0.29 μmol/L (Figure 1A), and its derivative carbenoxolone (a licensed drug) showed a slightly lower inhibitory activity with a Ki of 0.93 μmol/L (Figure 1B). Seven other natural pentacyclic triterpenes in this pool showed only slight mGLOI inhibitory activity (ursolic acid, oleanolic acid, betulic acid and tripterine with inhibition ratios <25% at a concentration of 10 μmol/L; Table 1) or no activity (maslinic acid, corosolic acid and madecassic acid; Table 1). Although a subsequent literature review indicated that GA had been identified as human GLOI inhibitor by Masayoshi et al in 198627, no further studies on the mechanism of inhibition or the structure-activity relationship were performed.

The Dixon plots and chemical structures of GA (A) and carbenoxolone (B). Data are the mean±SD (error bars) of n=3 experiments.
Structural basis for the inhibition of mGLOI by GA
To investigate the differences in GLOI inhibitory activity with the aforementioned pentacyclic triterpenes and to clarify the binding mode of GA, we determined the crystal structure of the mGLOI-GA complex at a 2.3 Å resolution. Similar to our previously determined complex structures of mGLOI-indomethacin and mGLOI-zopolrestat, the final structural model of mGLOI-GA also contained one GA molecule in one of the active sites, which was different from the other GLOI-inhibitor complexes, where two inhibitor molecules were found in both of the active sites of GLOI (PDB IDs: 1BH5, 1FRO, 1QIN, 1QIP, 2ZA0, 3VW9, 3W0T, and 3W0U). We also determined the mGLOI-GA complex structure in a different space group using a co-crystallization method and observed only one GA molecule per GLOI dimer at the asymmetric unit (unpublished data). Superimposition of both structures revealed no noticeable backbone movement or meaningful shift of the ligand position. Therefore, this phenomenon was very likely due to the method of preparation of the crystal of the enzyme-inhibitor complex, which was controlled by the affinity and solubility of the inhibitor.
A schematic presentation of the GA-occupied active site derived from the mGLOI-GA complex crystal structure revealed that GA did not coordinate with Zn2+; instead, it nestled in the GSH binding subpocket with its hydroxyl group pointing toward the glutamyl site and established a hydrogen-bonding network with mArg38B (-O2•••Nɛ, 2.4 Å; -O3•••HN, 3.2 Å), mAsn104B (-O2•••HN, 3.4 Å) and mArg123A (-O2•••HN, 3.2 Å; -O2•••HN, 3.4 Å) (Figure 2). The full occupation of GA in the GSH binding subpocket also resulted in extensive van der Waals interactions with the surrounding residues mMet36B, mMet66B, mPhe68B, mThr102B, mAsn104B, mVal150A, mLys151A, mPhe163A and mTrp171A, in addition to at least three nonpolar/polar and ten nonpolar/nonpolar contacts with distances shorter than 4 Å (Figure 2 and Table 2).

Structure of GA binding to mGLOI. 2Fo-Fc electron density map of GA contoured at 1.0 sigma. The protein and ligand surfaces are depicted as a transparent light blue and yellow cutaway. Key residues and ligands are shown as magenta and yellow stick models, respectively. Hydrogen bonds are shown as red dashed lines.
The binding mode of mGLOI-GA gave a rational explanation to the weak inhibitory activity of the other pentacyclic triterpenes tested here. For carbenoxolone, its flexible 3-O-hemisuccinate tail hung out of the active pocket and had few or very weak interactions with any residues of mGLOI predicted by the docking experiment (Supplementary Figure S1); therefore, carbenoxolone showed a similar inhibition as GA. As for ursolic acid, oleanolic acid, maslinic acid, corosolic acid, madecassic acid, and betulic acid, these six pentacyclic triterpenes showed little or no inhibition towards mGLOI due to their carboxyl groups located at the C17 position, which could not access either the glutamyl site or the glycyl site, as indicated by the structural superimposition with GA. For tripterine, the opposite stereochemistry of the carboxyl group on C30 to that of GA made it difficult to be anchored in the glutamyl site of the GSH binding site (Supplementary Figure S2). Thus, tripterine exhibited only slight inhibition of mGLOI.
Receptor model selection for a docking study with a non-GSH analog GLOI inhibitor
Notably, in silico GLOI inhibitor screenings published by our group and others have never identified GA as a possible candidate. In addition, our in silico screenings also did not identify zopolrestat or indomethacin as possible candidates. In fact, in our previously reported docking/MD study of the GLOI-indomethacin interaction mode, in which indomethacin coordinates with Zn2+ through its carboxyl group, a totally different result was revealed by the crystal structure19.
To investigate why GA was not identified in the in silico screening, we performed the docking studies with three representative GLOI-inhibitor crystal structures as receptors, ie, human GLOI-GIP (GSH/Zn2+-chelated inhibitor, the predominantly adopted receptor mode in in silico screening; PDB ID: 1QIN), mGLOI-MGI (non-GSH/Zn2+-chelated inhibitor; PDB ID: 2ZA0) and mGLOI-zopolrestat (non-GSH/non-Zn2+-chelation inhibitor; PDB ID: 4KYH). The results indicated that only with the GLOI-zopolrestat structure as the receptor did the docking study produce a similar conformation of GA to that observed in the crystal structure (Figure 3).

The binding conformations of GA derived from docking by employing crystal structure of human GLOI-GIP (PDB ID: 1QIN) (A), mGLOI-MGI (PDB ID: 2ZA0) (B) and mGLOI-zopolrestat (PDB ID: 4KYH) (C) as receptor, respectively. The crystal structure of GA derived from mGLOI-GA complex for comparison is shown as yellow sticks.
We compared the GLOI-ligand structures in the PDB database with that of apo mGLOI (unpublished data) and found that GIP and other GSH analog inhibitors induced a significant shift (>1.4 Å) of the glycyl site toward the active center, which did not occur in the non-GSH analog GLOI inhibitors or the apo structure (Supplementary Figure S3). Such a shift resulted in an obvious steric clash between the human Gly155A residue of the glycyl site and the methyl group at C4 of GA (1.2 Å) (Supplementary Figure S4). In contrast, such a conformational change was also likely to induce the docking programs to locate the ligands, such as indomethacin, at a deeper position in the active pocket to avoid steric interactions and therefore produce a false coordination between the ligands and Zn2+19. Because the docking method is critical for in silico screening and binding mode analysis, it is important to employ a suitable crystal complex structure as the receptor model when searching for non-GSH analog inhibitors.
Discussion
Since Vince et al proposed the concept of GLOI inhibitors as anti-tumor agents in the early 1970s28, GSH analogs with strong in vitro inhibition of GLOI have been extensively studied. However, because the GSH skeleton is very difficult to work with due to its pharmacokinetic properties5,10,11, non-GSH analog inhibitors could potentially lead to an alternative approach that yields a successful drug. Furthermore, considering the potential off-target effects of metal chelation20,21, non-GSH analog GLOI inhibitors lacking Zn2+-coordination may be more suitable for lead optimization in drug development. The discoveries of indomethacin, zopolrestat and GA as GLOI inhibitors and their related complex crystal structures have demonstrated the existence of a novel non-GSH/non-Zn2+-chelation type of GLOI inhibitor with a carboxyl group as an essential anchor to fix the inhibitors in the active site by hydrogen bonding. It is a reasonable conjecture that any compound with the ability to form strong hydrogen bonds with the glycyl site and/or the glutamyl site and the capability of fitting into the active pocket of GLOI could potentially be an effective GLOI inhibitor.
In addition to the GLO system, the aldose reductase (AR) is another important enzyme responsible for the detoxification of methylglyoxal and other α-oxoaldehydes29. A dual-target inhibitor that could simultaneously block GLOI and AR is likely to be more effective than a single-target GLOI inhibitor. GSH analog inhibitors are unlikely candidates for dual-target inhibitors because inhibition towards both GLOI and AR requires substituents with opposite polarities (hydrophobic and hydrophilic) on the thiol to bind to the active sites of GLOI and AR3,30. In fact, a carboxyl group is a typical (and critical) functional group for most known AR inhibitors31,32,33. Both indomethacin and zopolrestat are AR inhibitors with moderate (IC50=10 μmol/L)34 and strong (IC50=3 nmol/L)35 inhibition, respectively. Although GA is inactive towards AR36, it is a strong inhibitor of AKR1B10 (IC50 of 4.9 μmol/L)37, a close AR family member that also catalyzes detoxification of α-oxoaldehydes. Therefore, non-GSH/non-Zn2+-chelation type GLOI inhibitors containing a carboxyl group could have great potential as GLOI/AR dual-target inhibitors. Considering the large number of AR inhibitors synthesized since the 1990s, there is a strong possibility that there may be inhibitors with excellent in vivo anti-cancer activity in this group of compounds.
Author contribution
Xiao-peng HU and Qing LI designed the research; Hong ZHANG performed the experiments; Qiang HUANG, Jing ZHAI and Yun-yun CHEN assisted in the experiments; and Hong ZHANG wrote the manuscript with contribution from the other authors (Yi-ning ZHAO, Li-ping ZHANG, and Ren-wei ZHANG).
References
Thornalley PJ . Glyoxalase I--structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans 2003; 31: 1343–8.
Sousa Silva M, Gomes RA, Ferreira AE, Ponces Freire A, Cordeiro C . The glyoxalase pathway: The first hundred years and beyond. Biochem J 2013; 453: 1–15.
Cameron AD, Ridderström M, Olin B, Kavarana MJ, Creighton DJ, Mannervik B . Reaction mechanism of glyoxalase I explored by an X-ray crystallographic analysis of the human enzyme in complex with a transition state analogue. Biochemistry 1999; 38: 13480–90.
Kalsi A, Kavarana MJ, Lu T, Whalen DL, Hamilton DS, Creighton DJ . Role of hydrophobic interactions in binding S-(N-aryl/alkyl-N-hydroxycarbamoyl)glutathiones to the active site of the antitumor target enzyme glyoxalase I. J Med Chem 2000; 43: 3981–6.
Thornalley PJ, Rabbani N . Glyoxalase in tumourigenesis and multidrug resistance. Semin Cell Dev Biol 2011; 22: 318–25.
Creighton DJ, Zheng ZB, Holewinski R, Hamilton DS, Eiseman JL . Glyoxalase I inhibitors in cancer chemotherapy. Biochem Soc Trans 2003; 31: 1378–82.
Sousa Silva M, Ferreira AE, Gomes R, Tomás AM, Ponces Freire A, Cordeiro C . The glyoxalase pathway in protozoan parasites. Int J Med Microbiol 2012; 302: 225–9.
Suttisansanee U, Honek JF . Bacterial glyoxalase enzymes. Semin Cell Dev Biol 2011; 22: 285–92.
Vince R, Brownell J, Akella LB . Synthesis and activity of γ-(L-γ-azaglutamyl)-S-(p-bromobenzyl)-L-cysteinylglycine: a metabolically stable inhibitor of glyoxalase I. Bioorg Med Chem Lett 1999; 9: 853–6.
Takasawa R, Takahashi S, Saeki K, Sunaga S, Yoshimori A, Tanuma S . Structure-activity relationship of human GLO I inhibitory natural flavonoids and their growth inhibitory effects. Bioorg Med Chem 2008; 16: 3969–75.
Zhou Y, Guo T, Li X, Dong Y, Galatsis P, Johnson DS, et al. Discovery of selective 2,4-diaminopyrimidine-based photoaffinity probes for glyoxalase I. MedChemComm 2014; 5: 352–7.
Burg D, Mulder GJ . Glutathione conjugates and their synthetic derivatives as inhibitors of glutathione-dependent enzymes involved in cancer and drug resistance. Drug Metab Rev 2002; 34: 821–63.
Ramana KV, Bhatnagar A, Srivastava S, Yadav UC, Awasthi S, Awasthi YC, et al. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J Biol Chem 2006; 281: 17652–60.
Singh R, White MA, Ramana KV, Petrash JM, Watowich SJ, Bhatnagar A, et al. Structure of a glutathione conjugate bound to the active site of aldose reductase. Proteins 2006; 64: 101–10.
Kawatani M, Okumura H, Honda K, Kanoh N, Muroi M, Dohmae N, et al. The identification of an osteoclastogenesis inhibitor through the inhibition of glyoxalase I. Proc Natl Acad Sci U S A 2008; 105: 11691–6.
Chiba T, Ohwada J, Sakamoto H, Kobayashi T, Fukami TA, Irie M, et al. Design and evaluation of azaindole-substituted N-hydroxypyridones as glyoxalase I inhibitors. Bioorg Med Chem Lett 2012; 22: 7486–9.
Santel T, Pflug G, Hemdan NY, Schäfer A, Hollenbach M, Buchold M, et al. Curcumin inhibits glyoxalase 1— a possible link to its anti-inflammatory and anti-tumor activity. PLoS One 2008; 3: e3508.
Liu M, Yuan M, Luo M, Bu X, Luo HB, Hu X . Binding of curcumin with glyoxalase I: molecular docking, molecular dynamics simulations, and kinetics analysis. Biophys Chem 2010; 147: 28–34.
Liu M, Yuan M, Li Z, Cheng YK, Luo HB, Hu X . Structural investigation into the inhibitory mechanisms of indomethacin and its analogues towards human glyoxalase I. Bioorg Med Chem Lett 2011; 21: 4243–7.
Clutterbuck AL, Asplin KE, Harris P, Allaway D, Mobasheri A . Targeting matrix metalloproteinases in inflammatory conditions. Curr Drug Targets 2009; 10: 1245–54.
Jacobsen FE, Lewis JA, Cohen SM . The design of inhibitors for medicinally relevant metalloproteins. ChemMedChem 2007; 2: 152–71.
Zhai J, Zhang H, Zhang L, Zhao Y, Chen S, Chen Y, et al. Zopolrestat as a human glyoxalase I inhibitor and its structural basis. ChemMedChem 2013; 8: 1462–4.
Kawatani M, Okumura H, Honda K, Kanoh N, Muroi M, Dohmae N, et al. The identification of an osteoclastogenesis inhibitor through the inhibition of glyoxalase I. Proc Natl Acad Sci U S A 2008; 105: 11691–6.
Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994; 50: 760–3.
Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010; 66: 213–21.
Emsley P, Cowtan K . Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60: 2126–32.
Iio M, Ishimoto S, Nishida Y, Shiramizu T, Yunoki H . Effects of baicalein, a flavonoid, and other anti-inflammatory agents on glyoxalase-i activity. Agric Biol Chem 1986; 50: 1073.
Vince R, Daluge S, Wadd WB . Inhibition of glyoxalase I by S-substituted glutathiones. J Med Chem 1971; 14: 402–4.
Vander Jagt DL, Hunsaker LA . Methylglyoxal metabolism and diabetic complications: roles of aldose reductase, glyoxalase-I, betaine aldehyde dehydrogenase and 2-oxoaldehyde dehydrogenase. Chem Biol Interact 2003; 143–144: 341–51.
Steuber H, Czodrowski P, Sotriffer CA, Klebe G . Tracing changes in protonation: a prerequisite to factorize thermodynamic data of inhibitor binding to aldose reductase. J Mol Biol 2007; 373: 1305–20.
Hamada Y, Nakamura J . Clinical potential of aldose reductase inhibitors in diabetic neuropathy. Treat Endocrinol 2004; 3: 245–55.
Chung SS, Chung SK . Aldose reductase in diabetic microvascular complications. Curr Drug Targets 2005; 6: 475–86.
Obrosova IG, Kador PF . Aldose reductase/polyol inhibitors for diabetic retinopathy. Curr Pharm Biotechnol 2011; 12: 373–85.
Sharma YR, Cotlier E . Inhibition of lens and cataract aldose reductase by protein-bound anti-rheumatic drugs: salicylate, indomethacin, oxyphenbutazone, sulindac. Exp Eye Res 1982; 35: 21–7.
Wilson DK, Tarle I, Petrash JM, Quiocho FA . Refined 1.8 Å structure of human aldose reductase complexed with the potent inhibitor zopolrestat. Proc Natl Acad Sci U S A 1993; 90: 9847–51.
Lee YS, Kim SH, Jung SH, Kim JK, Pan CH, Lim SS . Aldose reductase inhibitory compounds from Glycyrrhiza uralensis. Biol Pharm Bull 2010; 33: 917–21.
Takemura M, Endo S, Matsunaga T, Soda M, Zhao HT, El-Kabbani O, et al. Selective Inhibition of the tumor marker Aldo-keto reductase family member 1B10 by oleanolic acid. J Nat Prod 2011; 74: 1201–6.
Acknowledgements
The authors thank Dr Hideo OKUMURA (Advanced Science Institute, RIKEN, Japan) for his kindly gift of the mouse GLOI plasmid. The authors gratefully thank the Innovative R&D Team Leadership of Guangdong Province, China (grant No 2011Y038).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available at Acta Pharmacologica Sinica's website.
Supplementary information
Supplementary Information
Supplementary infomration, Figures, Tables and Data (DOC 1372 kb)
Rights and permissions
About this article

Cite this article
Zhang, H., Huang, Q., Zhai, J. et al. Structural basis for 18-β-glycyrrhetinic acid as a novel non-GSH analog glyoxalase I inhibitor. Acta Pharmacol Sin 36, 1145–1150 (2015). https://doi.org/10.1038/aps.2015.59
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.59
Keywords
This article is cited by
-
New cytotoxic dammarane type saponins from Ziziphus spina-christi
Scientific Reports (2023)
