
Abstract
Aerobic glycolysis, also known as the Warburg effect, is a hallmark of cancer cell glucose metabolism and plays a crucial role in the activation of various types of immune cells. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) catalyzes the conversion of D-glyceraldehyde 3-phosphate to D-glycerate 1,3-bisphosphate in the 6th critical step in glycolysis. GAPDH exerts metabolic flux control during aerobic glycolysis and therefore is an attractive therapeutic target for cancer and autoimmune diseases. Recently, GAPDH inhibitors were reported to function through common suicide inactivation by covalent binding to the cysteine catalytic residue of GAPDH. Herein, by developing a high-throughput enzymatic screening assay, we discovered that the natural product 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose (PGG) is an inhibitor of GAPDH with Ki = 0.5 μM. PGG blocks GAPDH activity by a reversible and NAD+ and Pi competitive mechanism, suggesting that it represents a novel class of GAPDH inhibitors. In-depth hydrogen deuterium exchange mass spectrometry (HDX-MS) analysis revealed that PGG binds to a region that disrupts NAD+ and inorganic phosphate binding, resulting in a distal conformational change at the GAPDH tetramer interface. In addition, structural modeling analysis indicated that PGG probably reversibly binds to the center pocket of GAPDH. Moreover, PGG inhibits LPS-stimulated macrophage activation by specific downregulation of GAPDH-dependent glucose consumption and lactate production. In summary, PGG represents a novel class of GAPDH inhibitors that probably reversibly binds to the center pocket of GAPDH. Our study sheds new light on factors for designing a more potent and specific inhibitor of GAPDH for future therapeutic applications.
Similar content being viewed by others


Introduction
Glycolysis is a central glucose metabolism mechanism with metabolic pathways in the cytosol of all cells that generates the energy molecule ATP (adenosine triphosphate) and other intermediates. This multiple chemical reaction process can occur under anaerobic or aerobic conditions. Aerobic glycolysis was first described by Otto Warburg in early 1924 based on his research on cancer cell metabolism, which was accompanied by increased utilization of glucose without increasing oxygen consumption, even in a normoxic atmosphere [1, 2]. This phenomenon is not exclusive to cancer cells; many other normal cell types, for example, proliferating and quiescent fibroblasts and various immune cells (lymphocytes, natural killer cells, neutrophils, and macrophages), also exhibit a high aerobic glycolytic rate. Currently, considerable evidence has shown that both myeloid and lymphoid cells undergo a metabolic switch to aerobic glycolysis for supporting their functions [3, 4]. The glycolytic metabolic requirements of cancer cells and immune effect cells are different from those of normal cells and regulatory immune cells. Therefore, modulation of the aerobic glycolysis pathway may represent a potential therapeutic opportunity that may lead to the specific killing of tumor cells and suppression of excessively activated immune cells in cancer and autoimmune disease, respectively [5,6,7].
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (EC 1.2.1.12) is an NAD+-dependent oxidoreductase that converts glyceraldehyde 3-phosphate (GAP) to 1,3-bisphosphoglycerate (1,3-BPG) in the sixth step of glycolysis with an inorganic phosphate as a cofactor. Early studies defined GAPDH as a housekeeping gene due to its ubiquitous expression and abundance in various species [8,9,10]. Later, GAPDH was characterized as a moonlighting protein involved in a variety of cellular processes, including autophagy [11], apoptosis [12], cytoskeletal dynamics [13], DNA repair [14], tRNA transportation, and mRNA translation [15]. In addition, the overexpression and increased nuclear translocation of GAPDH in neurons are related to neuron death, which can be prevented by antisense knockdown of GAPDH [16]. GAPDH interacts with amyloid precursor protein and forms neurotoxic aggregates in the brains of patients with Alzheimer’s disease (AD) [17, 18], suggesting that GAPDH is associated with AD pathogenesis.
However, how GAPDH balances its versatile functions remains largely unclear. Considering metabolic control analysis (MCA) results, Liberti et al. [19] recently characterized GAPDH as a metabolic flux control enzyme under the Warburg effect and found that the partial inhibition of GAPDH by koningic acid (KA) was well tolerated and selective for highly glycolytic tumors. In addition, Chang et al. [20] demonstrated that GAPDH switches its glycolytic enzyme to IFNγ mRNA-binding protein during T cell activation, in which GAPDH was dissociated from the AU-rich element within the 3ʹ UTR of IFNγ to promote IFNγ secretion. Similarly, when macrophage activation is stimulated by LPS, cellular GAPDH is malonylated in the presence of malonyl-CoA and then dissociates from TNF mRNA to engage in glycolysis [21]. Furthermore, recent data suggest that dimethyl fumarate directly inactivates GAPDH, revealing a novel effective mechanism of the drug for use in multiple sclerosis treatment [22]. Thus, GAPDH has become a potential therapeutic target for heavily glycolytic tumors and autoimmune and neurological diseases [6, 23,24,25].
A few GAPDH inhibitors have been recently reported. These inhibitors are well-known molecules widely used in the clinic for Parkinson’s disease and autoimmune diseases (Fig. 1a). Selegiline, applied to treat movement disorders in Parkinson’s disease, was reported to block the S-nitrosylation of GAPDH, preventing its binding to Siah and thereby preventing cell death caused upon GAPDH-Siah complex nuclear translocation [16, 26]. However, selegiline is classified as a potent and irreversible inhibitor of monoamine oxidase A and B, which are critical for the degradation of dopamine, a central neurotransmitter in PD progression [27]. With respect to GAPDH, CGP 3466 (or TCH346) has a similar mechanism of action as selegiline, which disrupts the GAPDH-Siah interaction. This compound does not inhibit monoamine oxidase but offers more neuroprotection than selegiline. Its clinical development has been terminated due to lack of efficacy in PD and amyotrophic lateral sclerosis (ALS) patients [28]. KA, a fungal-derived natural product, acts as an irreversible inhibitor of GAPDH and selectively kills highly glycolytic cancer cells [19]. Dimethyl fumarate (DMF), a medication approved by the FDA for multiple sclerosis treatment, was discovered to be covalently linked to the catalytic cysteine residue of GAPDH to inactivate GAPDH enzymatic activity, leading to the downregulation of aerobic glycolysis in activated immune cells [22]. Notably, DMF also covalently modifies various other proteins, including Kelch-like ECH-associated protein 1 (KEAP1) [29] and gasdermin D (GSDMD) [30]. In general, although GAPDH has emerged as a potentially important therapeutic target, progress in developing potent and selective inhibitors is at an early stage. Therefore, we sought to discover novel GAPDH inhibitors as valuable chemical tools for probing GAPDH functions and disease-associated biology.

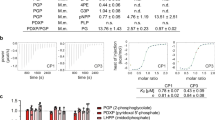
a Reported inhibitors of GAPDH. b Principles of the high-throughput screening assay. Na2AsO4 was introduced to replace PGK1 and eliminate the production inhibition of GAPDH and reduce the false positive hit rate. WST/1-mPMS was introduced to improve assay sensitivity. c Parameters for evaluating the quality of our designed high-throughput screening assay. Groups with or without GAPDH as positive or negative controls were assessed to determine the assay parameters. The Z’ factor, signal-to-noise ratio (S/N), and signal-to-background were calculated by the method described in Reference [37]. d The workflow of the compound library screening. e Chemical structure of PGG, and the IC50 for GAPDH = 6.7 ± 0.98 μM.
Herein, we developed a robust, sensitive, and cost-effective in vitro high-throughput enzymatic assay for screening GAPDH inhibitors. In addition, several molecules that show inhibitory activities against GAPDH were successfully identified. Among these inhibitors, the most promising is a natural product named 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose (PGG) [31, 32]. Kinetic analysis demonstrated that PGG is a reversible competitive inhibitor of GAPDH. HDX-MS revealed that PGG binds to two critical fragments and significantly enhances the deuterium uptake rate at the tetramer interface of GAPDH. Structural modeling analysis suggested that PGG probably binds to the center pocket of GAPDH. Furthermore, we demonstrated that PGG can target cellular GAPDH to suppress the glycolysis pathway and inhibit LPS-stimulated macrophage activation, suggesting that PGG can modulate GAPDH target-specific cellular functions. In summary, we discovered that the natural product PGG is a novel inhibitor of GAPDH and elucidated its potential GAPDH inhibition mechanism. The unique mechanism of action of PGG, which involves binding to the center pocket of GAPDH overlapping with NAD+ and Pi sites, sheds light on designing and optimizing more-potent and specific inhibitors of GAPDH for probing its functions and using it in therapeutic applications in the future.
Materials and methods
Protein expression and purification
The DNA fragment encoding the full-length human GAPDH gene was amplified from the complementary DNA library of 293 T cells and subcloned into a pET-28a vector incorporating an N-terminal TEV (tobacco etch virus)-cleavable His6 tag. The expression plasmid was verified by Sanger sequencing. The corresponding plasmid was transformed into BL21 (DE3) PlysS Escherichia coli cells (TransGen Biotech), cultured in 1 L of Luria-Bertani (LB) medium at 37 °C until the absorbance at OD600 was 0.4–0.6, and then, the cells were induced with 0.4 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) and cultured overnight at 16 °C. The cells were harvested by centrifugation and stored at −80 °C until further use. To purify human GAPDH proteins, the pellets were resuspended and lysed in lysis buffer (20 mM HEPES, pH 7.4; 200 mM NaCl; 4 mM β-mercaptoethanol; and 20 mM imidazole) and centrifuged at 16,000 × g for 1 h at 4 °C. The supernatant was loaded into a nickel affinity column (GE Healthcare) with a peristaltic pump. Proteins were eluted with gradient imidazole and assessed by SDS-PAGE electrophoresis. Fractions containing GAPDH and with a purity greater than 90% were collected and digested with TEV protease for 16 h at 4 °C and then passed over a nickel affinity column (GE Healthcare) to remove cleavage tags. The remaining fraction was further purified via size-exclusion chromatography by a Superdex 200 gel filtration column (GE Healthcare) equilibrated in gel filtration buffer (20 mM HEPES, pH 7.4; 200 mM NaCl; and 1 mM dithiothreitol). Fractions containing GAPDH were concentrated to 10 mg/mL, aliquoted and stored at −80 °C in storage buffer (20 mM HEPES, pH 7.4; 200 mM NaCl; and 1 mM DTT) with liquid nitrogen until further use.
Assay development and optimization
All kinetic assays were performed in assay buffer (100 mM HEPES, pH 7.4; 100 mM NaCl; 2 mM MgCl2; 0.1 mM DTT; 0.5 mM EDTA; and 1 mg/mL BSA) and monitored by spectrometry in kinetics mode. A 96-well format assay with a total volume of 200 µL was used to determine cell lysate GAPDH activity not coupled to water-soluble tetrazolium-8 (WST-8)/ 1-methoxy-5-methylphenazium methyl sulfate (1-mPMS) reagent, and a Z’ factor of a 384-well format assay with 40 µL total volume was performed in the absence or absence of WST/1-mPMS reagent. The 384-well format assay coupled with WST/1-mPMS was applied to high-throughput screening and evaluation of compound potency.
The Michaelis constant (Km) value was assayed in a buffer at final volume of 40 µL (100 mM HEPES, pH 7.4;100 mM NaCl; 2 mM MgCl2; 0.1 mM DTT; 0.5 mM EDTA; and 1 mg/mL BSA) in a 384-well format. A series of different substrate concentrations (NAD+ from 0 mM to 5 mM, D-GAP from 0 mM to 2.5 mM, Na2AsO4 from 0 mM to 60 mM, Na2HPO4 from 0 mM to 25 mM) was prepared, and the enzyme reaction was initialized by adding GAPDH protein (final concentration of 10 nM). The final concentration of WST-8 was 1 mM, and that of 1-mPMS was 0.04 mM. All kinetic assays were performed in the same assay buffer with consensus concentrations of enzyme and detection reagent at initial velocity. The initial velocity was measured when <10% of products had formed. The same series of substrate concentrations without any enzyme were also measured as a control, and the buffer without protein or substrates was measured to serve as the background control. Absorbance intensity (the WST/1-mPMS reagent group at 450 nm, and the NADH without the WST/1-mPMS reagent group at 340 nm) was monitored continuously for 15 min with a microplate reader (Thermo Fisher) at 37 °C. The Michaelis constant (Km) and maximum activity (Vmax) were calculated by fitting the data to a Michaelis-Menten equation (Km): [33]
The Km for GAP was determined using a concentration range of 0–2.5 mM at a fixed NAD+ concentration of 1 mM and fixed Na2AsO4 concentration of 20 mM. The Km for NAD+ was measured using a concentration range of 0–5 mM at a fixed GAP concentration of 2 mM and fixed Na2AsO4 concentration of 20 mM. The Km for Na2AsO4 was measured using a concentration range of 0–60 mM at a fixed GAP concentration of 2 mM and fixed NAD+ concentration of 1 mM. The Km for Na2HPO4 was measured using a concentration range of 0–25 mM at a fixed GAP concentration of 2 mM and fixed NAD+ concentration of 1 mM. GraphPad Prism 6.0 software was used to calculate the values of Km by fitting the data to the Michaelis-Menten equation. The data can be found in Supplementary Fig. S1.
To further validate this assay, 50 mM solution of the positive compound named Heptelidic acid (also Koningic acid) (EFEBIO) was prepared in 100% DMSO as stock solution, diluted to various concentrations with assay buffer and incubated with 10 nM GAPDH protein for 15 min. The reaction was initiated by adding substrate buffer (1 mM NAD+, 2 mM D-GAP, and 5 mM Na2AsO4). The absorbance intensity was then measured by the same method as used to measure the Km.
Primary high-throughput screening
Substrate concentrations are vital for enzyme high-throughput assays for the discovery of different mechanisms of action inhibitors. Considering the Km of each substrate, initial velocity conditions, and the robustness of the high-throughput assay, we used 2 mM D-GAP (Km = 149.0 ± 42.7 μM), 1 mM NAD+ (Km = 148 ± 17.8 μM), 5 mM Na2AsO4 (Km = 15.2 ± 1.7 mM), and 10 nM GAPDH for the high-throughput screening assay. The same conditions were also used to calculate the IC50 of the hit compounds. We used an in-house natural product library for HTS. The in-house natural product collection included 2000 compounds. All compounds were stored as 10 mM stock solutions dissolved in 100% DMSO under drying conditions at −20 °C. All assays were performed in 384-well plates (Corning) at room temperature. The GAPDH protein enzyme (10 nM final concentration) was prepared in assay buffer. Twenty-two microliters of enzyme buffer was dispensed into wells, and 200 nL of 10 mM compound (50 µM final concentration of each) was added and shaken evenly with a vibrator at 800 × g. After incubating for 15 min, enzyme reactions were initiated by adding 18 µL of the substrate solution (1 mM NAD+, 2 mM D-GAP, 5 mM Na2AsO4, 1 mM WST-8, and 0.04 mM 1-mPMS as the final concentration). Steady-state reactions were recorded on a microplate reader at 450 nm for 15 min, with readings taken every 30 s.
Hit confirmation and IC50 determination
All hit compounds from the HTS were selected and reanalyzed by manual continuous kinetic analysis for confirmation. Compounds with increased signal in the absence of GAPDH, indicating false positive hits, were excluded. For compound that inhibition of >40% in the confirmation assay, IC50 values were measured in triplicate using the same assay conditions as the initial screening. In 384-well plates, 12 µL of enzyme solution was distributed into wells, and 10 µL of different concentrations of compounds were added and then incubated for 15 min. The enzyme reaction was initiated by adding 10 µL of the substrate buffer and 8 µL of WST/1-mPMS reagent buffer. The IC50 values were obtained by fitting data to a four-parameter dose-response curve in GraphPad Prism 6 software.
One-dimensional NMR spectroscopy
Ligand-observed CPMG and saturation transfer difference (STD) NMR experiments were performed to investigate ligand-protein interactions. All NMR spectra were acquired at 25 °C on a Bruker Avance III 600 MHz NMR spectrometer equipped with a cryoprobe (Bruker Bio-Spin, Germany). Samples containing 200 μM PGG and 200 μM PGG in the presence of 10 μM GAPDH were dissolved in phosphate buffer (20 mM NaH2PO4, 20 mM Na2HPO4, 100 mM NaCl, 95% D2O, and 5% DMSO-d6, pH 7.4).
Microscale thermophoresis
Recombinant GAPDH was labeled with Monolith protein labeling kit RED-NHS 2nd Generation (Cat# MO-L011) according to the labeling protocol provided by the manufacturer. Labeled proteins were used at a concentration of 10 nM. Samples were diluted in 20 mM HEPES (pH 8.0) and 0.05 (v/v) Tween-80. We used 500 µM PGG as the highest concentration for the serial dilution. After 10 min of incubation at 4 °C and centrifugation at 12,000 × g, the samples were loaded into Monolith standard-treated capillaries, and then, thermophoresis was measured on a Monolith NT automated (MA-052) instrument (NanoTemper). Laser power was set to 20% using a Xs on time. The LED power was set to auto. The dissociation constant (Kd) values were fitted by using NTAnalysis software (NanoTemper Technologies, München, Germany).
Recovery of enzyme activity after dilution
PGG (IC50 of GAPDH = 7 µM) and KA (IC50 of GAPDH = 40 nM) at concentrations equal to 10 × IC50 (60 µM) for the inhibition of GAPDH were preincubated with recombinant human GAPDH (200 nM) for 30 min at room temperature in enzyme assay buffer (100 mM HEPES (pH 7.4), 100 mM NaCl, 2 mM MgCl2, 1 mM DTT, 0.5 mM EDTA, and 1 mg/mL BSA). A control incubation was performed in the absence of PGG, and DMSO (0.5%) was added as a cosolvent to all preincubated compounds and the control. The reactions were diluted 20-fold with enzyme assay buffer to obtain a final inhibitor concentration equal to 0.5 × IC50, and the final concentration of GAPDH was still 10 nM. The diluted 0.5 × IC50 PGG (3 µM), KA (20 nM), and GAPDH protein was then incubated at room temperature for another 20 min, substrate buffer and WST/1-mPMS reagent buffer were added, and finally, the absorbance at 450 nm was measured at 25 °C for 15 min.
Hydrogen-deuterium exchange (HDX)
Hydrogen-deuterium exchange (HDX) mass spectrometry (MS) for peptide identification
Peptides were identified using tandem MS (MS/MS) with a Fusion Orbitrap mass spectrometer (Thermo Fisher). Product ion spectra were acquired in data-dependent mode with the top eight most abundant ions selected for the product ion analysis per scan event. The MS/MS data files were entered into Proteome Discoverer 2.4 (Thermo Fisher) for high-confidence peptide identification.
HDX-MS analysis
Five microliters of GAPDH (50 mM HEPES, pH 7.4; 150 mM NaCl; 5% glycerol; 5 mM MgCl2; and 2 mM DTT) was incubated with and without the compound at a 1:20 molar ratio (protein:ligand) for 0.5 h before the HDX reacted at 4 °C. Four microliters of protein/protein complex with ligand/peptide was diluted into 16 µL of D2O in exchange buffer (50 mM HEPES, pH 7.4; 50 mM NaCl; and 2 mM DTT) and incubated for various HDX time points (e.g., 0, 10, 60, 300, and 900 s) at 4 °C and quenched by mixing with 20 µL of ice-cold 3 M guanidine hydrochloride and 1% trifluoroacetic acid. Each quenched sample was immediately injected into the LEAP Pal 3.0 HDX platform. Upon injection, samples were passed through an immobilized pepsin column (2 mm × 2 cm) at 120 µL/min, and the digested peptides were captured on a C18 PepMap300 trap column (Thermo Fisher) and desalted. Peptides were separated with a 2.1 mm × 5 cm C18 separating column (1.9 μm Hypersil Gold, Thermo Fisher) with a linear gradient of 4–40% CH3CN and 0.3% formic acid over 6 min. Sample handling, protein digestion, and peptide separation were conducted at 4 °C. Mass spectrometric data were acquired using a Fusion Orbitrap mass spectrometer (Thermo Fisher) with a measured resolving power of 65,000 at m/z 400. HDX analyses were performed in triplicate for each preparation of a single protein-ligand complex. The intensity weighted mean m/z centroid value of each peptide envelope was calculated and subsequently converted into a percentage of deuterium incorporation. Statistical significance for the differential HDX data is determined by an unpaired t test for each time point, a procedure that is integrated into the HDX Workbench software [34]. Corrections for back exchange were made on the basis of an estimated 70% deuterium recovery and accounting for the known 80% deuterium content of the deuterium exchange buffer.
Data rendering
The HDX data from all overlapping peptides were consolidated to individual amino acid values using a residue averaging approach. Briefly, for each residue, the deuterium incorporation values and peptide lengths from all overlapping peptides were assembled. A weighting function was applied in which shorter peptides were weighted more heavily and longer peptides were weighted less heavily. Each of the weighted deuterium incorporation values were then averaged to produce a single value for each amino acid. The initial two residues of each peptide, as well as proline residues, were omitted from the calculations.
Structural modeling
The docking pose of PGG in GAPDH was generated by using Schrödinger software. Ligand preparation was used to generate different conformations of PGG. E. coli-activated GAPDH (PDB ID: 1DC4) and Bacillus stearothermophilus GAPDH (PDB ID: 1NQO) bound to both GAP and NAD+. Human GAPDH with NAD+ binding (PDB ID: 1U8F) was used to generate the docking grid file by Receptor Grid generation with default parameters. Glide was used for ligand docking. Vacuum electrostatic views of GAPDH and modeled PGG were generated by PyMOL.
Cell culture and reagents
RAW264.7 and HCT116 cells were purchased from ATCC (American Type Culture Collection). The cells were cultured in DMEM supplemented with 10% FBS (fetal bovine serum) and 1% PS (penicillin/streptomycin) at 37 °C in a 5% CO2 atmosphere. Anti-GAPDH antibody (CST, cat #5174 s), anti-HSP90 (CST, cat #4877 T), anti-iNOS (Abcam, ab178945), anti-cleaved-IL-1β (Asp117) (CST, cat#63124) were used in this study. GAPDH small interfering RNA was synthesized by Gene Pharm (GM). LPS (E. coli, 055:B5, Sigma) and D-GalN (Sigma) were purchased from Sigma, and they were dissolved in PBS for use. WST-8 and 1-mPMS were purchased from GLPBio. Glucose and lactate analyzer kits were purchased from NICHIWA Trading Co., Ltd.
Dynamic light scattering assay
GAPDH (5 μM) was incubated with 100 μM PGG or 0.2% DMSO for 5 min and centrifuged at 13,000 rpm for 15 min at 4 °C. The supernatants were subjected to DynaPro Plate Reader (WYATT). Each point has five replications.
Small interfering RNA transfection assay
RAW264.7 cells (500,000) were plated in 12-well culture plates to allow cell attachment overnight, and then, Lipofectamine RNAiMax was used for transfection. A total of 80 pmol control siRNA and three GAPDH siRNAs were transfected into each well according to the manufacturer’s recommended procedure. The cells were harvested at 48 h after transfection for Western blot and RT-qPCR analyses.
Quantitative RT-PCR experiment
Cells were treated as indicated and then harvested for RT-qPCR analysis. A FastPure Cell/Tissue Total RNA isolation kit (Vazyme Biotech, RC101) was used to extract total RNA from the cells, and this RNA was reverse transcribed into cDNA using a HiScript Q RT SuperMix kit (Vazyme Biotech, R122-01). ChamQ SYBR qPCR master mix (Vazyme Biotech, Q331-02) was used for qRT-PCR analysis with a Quant Studio 6 Flex real-time PCR system (Applied Biosystems). All experiments were performed according to the manufacturer’s instructions (Vazyme Biotech). All primers were listed in Table 1.
Glucose and lactate level determination
RAW264.7 cells (1.5 × 106) were plated into each well of a 6-well plate in basic DMEM containing 25 mM glucose, 2 mM glutamine, and 10% dialyzed FBS (Gibco) (cultured media). The cells were allowed to attach overnight. After pretreatment with the indicated concentrations of compounds in fresh culture medium for 2 h, 1 μg/mL LPS was added to stimulate the cells for another 10 h. The supernatant medium was collected for glucose and lactate measurements. The remaining glucose in the cell culture medium was measured by the hexokinase/glucose-6 phosphate dehydrogenase method. The lactate concentration in the cell culture medium was measured by the lactate dehydrogenase method. All samples were prepared with commercial assay kits and analyzed in an automatic analyzer (HiTACHI 7020).
Animals and treatment
Six- to eight-week-old male C57BL/6 J mice were purchased from SIMM Animal Center (Shanghai, China) and maintained under standard laboratory conditions. All animal experiments were approved by the Institute Animal Care and Use Committee at the Shanghai Institute of Materia Medica. Thirty mice weighing 21–23 g were randomly divided into three groups (n = 10): normal group, model group, and treatment group. Normal group and model group mice were pre-orally administered vehicle (1% DMSO in PBS), and the treated group mice were pre-orally administered 60 mg/kg PGG. Two hours later, the normal group mice were intraperitoneally injected with PBS, the model group mice were intraperitoneally injected with LPS (2 mg/kg) and D-GalN (250 mg/kg), and the treatment group mice were intraperitoneally injected with 2 mg/kg LPS and 250 mg/kg GalN. The survival rate was recorded every 2 h for 24 h.
Quantification and statistical analysis
All numerical results are expressed as the mean ± SEM representing three independent experiments or three parallel experiments. All analyses were performed with GraphPad Prism 6.0 software. Two-tailed unpaired Student’s t test was used to analyze differences between the two groups. The significance level was set at *P < 0.05; **P < 0.01, ***P < 0.001, ****P < 0.0001.
Results
Screening and identification of PGG as an inhibitor of GAPDH
GAPDH catalyzes glyceraldehyde-3-phosphate (GAP) to 1,3-biphosphoglycerate (1,3-BPG) with the concomitant reduction of NAD+ to NADH. The free-energy change of the GAPDH catalytic reaction is unfavorable, but cells overcome this by the rapid removal of 1,3-BPG in a favorable subsequent step catalyzed by phosphoglycerate kinase (PGK). Therefore, the conventional method for the detection of GAPDH enzymatic activity is usually coupled with phosphoglycerate kinase. This assay system is not ideal for high-throughput screening due to difficulties in dissecting PGK interference from true GAPDH inhibition. To overcome this disadvantage, we designed a new high-throughput GAPDH enzymatic assay. First, we substituted arsenate for inorganic phosphate to eliminate production inhibition of GAPDH. In the presence of arsenate, GAPDH converts glyceraldehyde-3-phosphate to 1-arseno-3-phosphoglycerate, which is unstable and is rapidly hydrolyzed to 3-phosphoglycerate (3-PG) and hydrogen arsenate [35], thus allowing for a favorable equilibrium for GAPDH rate activity in the forward direction (Fig. 1b). This reaction continues without intervention by phosphoglycerate kinase and therefore can reduce the false positive hit rate. Second, WST-8 was introduced to the assay system to improve assay sensitivity, and importantly, its product can serve as a reaction detection molecule. Tetrazolium salts can be converted to water-soluble formazan in the presence of NADH. WST-8 together with the electron mediator 1-mPMS, commercially known as CCK-8, is a reagent that has been widely used in the measurement of cellular NAD(P)H metabolite concentration as an indicator of cell viability [36]. WST-8 is reduced by NADH to form a strong orange formazan product that shows maximal absorption at 450 nm. In the presence of WST-8/1-mPMS, NADH generated by GAPDH is consumed in real time to convert WST-8 to WST-8 formazan. The absorbance of formazan is proportional to the GAPDH-produced NADH concentration and therefore can be used to monitor GAPDH activities by spectrophotometry in a rapid, accurate, and cost-effective fashion. Finally, we scaled the assay system into a 384-well plate format that requires less GAP reagent for improved automation efficiency.
The sensitivity and limitation of the detection were determined by the standard curve of NADH in the presence of WST/1-mPMS. The determination of all substrate concentrations depended on the Km of each substrate, and the initial velocity was measured as less than or equal to 10% of the product formed. Details of this assay can be found in “Materials and methods” section. The high-throughput screening assay showed good quality with a Z’-factor of 0.71 [37] (Fig. 1c). To further assess the reliability and robustness of the assay, we measured the inhibitory activity of KA on GAPDH. We found that KA had an IC50 of 39 nM in our assay, which is consistent with the reported results (Supplementary Fig. S2). Overall, we developed and optimized a new GAPDH high-throughput assay for compound screening.
Natural products and their derivatives are important sources of effective drug candidates. Indeed, many natural products have been approved for treating diseases, including cancer, autoimmune, and infectious diseases [38]. We have gained experience in identifying natural product-based drug candidates for a considerable number of targets [39,40,41,42]. Hence, we applied this new high-throughput assay to screen an in-house focused natural product library containing 2000 compounds to discover novel GAPDH inhibitors. The workflow for finding hits is shown in Fig. 1d. Briefly, an enzyme solution was preincubated with compounds for 10 min, and a substrate solution (including NAD+, GAP, and WST-8/1-mPMS) was then added and mixed to initiate the reaction. The absorbance at 450 nm was monitored in continuous kinetic mode for 15 min. All compounds were tested at a single dose of 50 μM in the primary screening. Compounds with inhibition activity greater than 50% compared to the DMSO control were selected for a second screening. The selected hit compounds were rescreened in three doses in triple replicates. Compounds that had intrinsic absorbance at 450 nm or showed no concentration response were eliminated. The IC50 values of the remaining compounds were determined in a dose-dependent manner to rank their relative GAPDH inhibition activities. Among these candidates, 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose (PGG) was identified as the most potent compound, with an IC50 of 6.7 ± 0.98 μM (Fig. 1e). PGG is a galloyl-beta-D-glucose compound with five galloyl groups; therefore, we tested whether gallic acids have inhibitory activity against GAPDH. However, gallic acids showed no inhibitory activity against GAPDH at 200 μM (data not shown). PGG is a tannin family compound and is highly enriched in several natural plants, especially traditional herbals. Studies have shown that PGG has multiple activities, including antimicrobial, anti-inflammatory, anticarcinogenic, antidiabetic, antitumor, and antioxidant activities; [43,44,45] therefore, PGG inhibition of GAPDH activity is viewed favorably.
PGG is a reversible and competitive inhibitor of GAPDH
To rule out the possibility of nonspecific binding of PGG to GAPDH, ligand-based nuclear magnetic resonance (NMR) spectroscopy methods were applied to validate the PGG-GAPDH interaction. Carr-Purcell-Meiboom-Gill (CPMG) together with saturation transferred difference (STD) clearly revealed specific binding of PGG to GAPDH (Supplementary Fig. S3a and S3b). A microscale thermophoresis (MST) assay was then used to determine the binding affinity of PGG for GAPDH, and the results showed a Kd of ~14.5 μM (Fig. 2a). Our data confirmed that PGG specifically binds GAPDH.

a The binding affinity of PGG for GAPDH as determined by microscale thermophoresis (MST) assay. The data represent three independent experiments, and the error bars show the SEM. b V0-Enzyme plot with fixed doses of inhibitors (KA = 0.5 μM and PGG = 100 μM) showed that PGG is a reversible inhibitor. The data were generated in triplicate, and the error bars represent the standard error of the mean (SEM). c–f Steady-state enzyme kinetic assay shows kinetic parameters with various concentrations of PGG with respect to GAP (c), NAD+ (d), Pi (e), and Na2AsO4 (f). All steady-state assays were performed with a 10 nM enzyme concentration. The Km for GAP was determined using a concentration range of 0–4 mM at a fixed NAD+ concentration of 1 mM and fixed Na2AsO4 concentration of 20 mM with the indicated concentration of PGG. The data were fitted by GraphPad Prism software in noncompetitive mode. The Km for NAD+ was measured using a concentration range of 0–4 mM at a fixed GAP concentration of 2 mM and fixed Na2AsO4 concentration of 20 mM with the indicated concentration of PGG. The data were fitted by GraphPad Prism software in competitive mode. The Km for Na2HPO4 was measured using a concentration range of 0–20 mM at a fixed GAP concentration of 2 mM and fixed NAD+ concentration of 1 mM with the indicated concentration of PGG. The data were fitted by GraphPad Prism software in competitive mode. The Km for Na2AsO4 was measured using a concentration range of 0–40 mM at a fixed GAP concentration of 2 mM and fixed NAD+ concentration of 1 mM with the indicated concentration of PGG. The data were fitted by GraphPad Prism software in competitive mode. The data represent three parallel experiments, and the error bars represent the SEM.
Next, we aimed to understand the enzymatic mechanism of PGG inhibition of GAPDH. First, steady-state enzyme kinetics analysis unambiguously revealed that PGG is a reversible inhibitor of GAPDH (Fig. 2b). The inhibitor KA exhibited a shifted linear curve between Vmax and the enzyme concentration with a similar catalytic slope to that of inhibitor-free GAPDH. In contrast, GAPDH inhibition by PGG showed a linear but reduced catalytic slope (Fig. 2b). The results clearly indicate that KA is an irreversible inhibitor of GAPDH, as its inhibition led to the similar increased initial velocity as that of the free GAPDH. In contrast to KA, a kinetic analysis unambiguously revealed that PGG is a reversible inhibitor of GAPDH since it showed a reduced initial velocity, a phenomenon typical of dissociation between the enzyme and inhibitor complex during the reaction. The reversible inhibition property of PGG was also revealed by the recovery of enzyme activity after dilution of the enzyme-inhibitor mixture, as the enzymatic activity of GAPDH was recovered when the GAPDH-PGG complex concentration was diluted from 10 × IC50 to 0.5 × IC50. In contrast, after similar treatment of GAPDH with KA, the GAPDH enzymatic activity was not recovered (Supplementary Fig. S3c and S3d).
We then conducted classical steady-state experiments to further understand the binding mode and potency of PGG as an inhibitor of GAPDH. Under fixed and saturated concentrations of NAD+ and Na2AsO4, the Km of the substrate GAP did not change, while the Vmax of GAP decreased with increased PGG, suggesting that PGG does not compete with GAP (Fig. 2c). However, under fixed and saturated concentrations of GAP and Na2AsO4, the Vmax of NAD+ showed no obvious change, while the Km of NAD+ increased with increasing concentrations of PGG, suggesting that PGG competes with NAD+ (Fig. 2d). Inorganic phosphate (Pi) is also a cofactor important for GAPDH catalysis; thus, we evaluated the kinetic parameters of Pi and arsenate under fixed and saturated concentrations of GAP and NAD+. The Vmax of Pi and arsenate showed no obvious change, while the Km of Pi and arsenate increased with increasing concentrations of PGG, suggesting that PGG competes with Pi (Fig. 2e) or arsenate (Fig. 2f). All kinetic parameters are listed in a table of Fig. 2. The non-competitive inhibition nature of PGG with respect to GAP demonstrates that GAP is not involved in nor affects PGG binding. The competitive mode of PGG with respect to NAD+ and Pi indicates that PGG prefers to bind to free enzymes over the Michaelis complexes formed by NAD+-GAPDH, Pi-GAPDH or Pi-GAPDH-NAD+, with a high potency of Ki = 0.48 ± 0.01 μM. Taken together, PGG is a reversible and NAD+ and Pi competitive inhibitor of GAPDH.
PGG binds to the center pocket of GAPDH, as revealed by HDX-MS analysis and structural modeling
Since PGG competes with NAD+, we speculated that PGG probably occupies the NAD+ pocket. However, neither structure of PGG is similar to NAD+, nor is the cavity of NAD+ pocket large enough to accommodate PGG binding. The cavity of NAD+ is an extended conformational groove site, while the shape of PGG is a stereoscopic conformation that is clearly incompatible to accommodate the NAD+ pocket. Thus, we hypothesized that PGG may bind to different sites and allosterically prevent NAD+ from binding to GAPDH.
We attempted to resolve the crystal structure of GAPDH in complex with PGG, but unfortunately, we could not obtain useful crystals with good diffraction. We then turned to conformational dynamics studies by hydrogen deuterium exchange mass spectrometry (HDX-MS), a well-established technology that has been widely applied to study protein structures, dynamics, folding, and interactions [46, 47]. The different deuterium uptake rates of GAPDH upon PGG binding (Supplementary Fig. S4a and S4b) revealed the specific regions that interact with PGG as well as conformational changes induced by PGG binding. Four regions showed significantly different deuterium incorporation in PGG-treated GAPDH compared to control GAPDH. Interestingly, region I, covering residues 96–102, showed an average 15% decrease in deuterium uptake, and region II, covering residues 206–218, showed an approximate 10% decrease in deuterium uptake (Fig. 3a). Regions III (residues 176–204) and IV (residues 278–286) exhibited an approximate 10% increase in deuterium uptake upon GAPDH binding to PGG (Fig. 3b). The HDX-MS results suggest that these regions probably serve as important elements in PGG binding.

a The deuterium uptake plot for region I and region II are shown. Deuterium uptake plot of the peptide VVESTGVF (AA 96–103, +1 charge state) (region I) and QNIIPASTGAAKAVG (AA 205–219, +2 charge state) (region II) in GAPDH in the presence and absence of PGG are shown. The data are plotted as percent deuterium uptake versus time on a logarithmic scale. Red and blue plots represent the GAPDH and the PGG bound state, respectively. b The deuterium uptake plot for region III and region IV are shown. Deuterium uptake plot of the peptide TTVHAITATQKTVDGPSGKL (AA 177–196 +4) (region III) and HQVVSSDF (AA 280–287 +2) (region IV) for GAPDH in the presence and absence of PGG are shown. The data were plotted as percent deuterium uptake versus time on a logarithmic scale. Red and blue plots represent the GAPDH and the PGG bound state, respectively. c Differential HDX consolidation view (related to Supplementary Fig. S3A and S3B) was mapped to + the GAPDH crystal structure (PDB ID: 1U8F). The GAPDH monomer is shown on the right, the enlarged view was also modeled with GAP and NAD+, NAD+ was modeled by 1U8F, the modeled GAP was based on the PDB ID: 1NQO of Bacillus stearothermophilus GAPDH. HDX Workbench colors each peptide according to the smooth color gradient HDX perturbation key (D%), as shown in each figure. Differences in %D between −5% and 5% are considered non-significant and are colored gray according to the HDX perturbation key shown below. In addition to the −5% to 5% test, unpaired t tests were calculated to determine significant differences (P < 0.05) between samples at each time point. A negative value represents decreased deuterium incorporation or stabilization, while a positive value represents increased deuterium incorporation or destabilization in the corresponding region of the receptor when a binding event takes place. Peptides exhibiting statistically insignificant or undetectable changes are colored gray. The blank region represents undetected peptide for the corresponding experiment.
To understand the potential molecular mechanism of PGG inhibition, these amino acid regions were then mapped onto the crystal structure of GAPDH (PDB ID: 1U8F) (Fig. 3c) of the modeled substrate GAP and cofactor NAD+. Since the HDX-MS assay was performed in the basal state of human GAPDH, NAD+ was modeled in the basal state of the human GAPDH structure (PDB ID: 1U8F), and GAP was modeled in the basal state of Bacillus stearothermophilus GAPDH (PDB ID: 1NQO). Region I is located at the N-terminal domain of GAPDH, region II is in the C-terminal domain of GAPDH, and both regions III and IV are located at the tetramerization interface of GAPDH (Fig. 3c, left magnified). Then, using structural modeling, we tried to locate the potential site that accommodates PGG binding. Intriguingly, the mapped structure indicates a large pocket consisting of region I, region II, and region III possibly suitable for PGG binding. This pocket is composed of the inorganic phosphate site, C3-phosphate (C3P) group of the GAP binding site (Ps site), and nicotinamide moiety of NAD+. We termed this pocket the center pocket because it crosses the N-terminal domain and the C-terminal domain. Specifically, five amino acids in region I are among the residues critical for interaction with the cofactor NAD+ (Fig. 4a, right magnified). Glu97 and Gly100 form water-mediated interactions with nicotinamide sugars. Ser98 forms water-mediated polar interactions with pyrophosphate, and Thr99 and Phe102 form nonpolar interactions with adenine. The decrease in deuterium uptake in region I may be attributed to the displacement of NAD+ by PGG binding to GAPDH. In addition, residues Thr211 and Gly212 residing in region II coordinate with the catalytic center residue S151 to form the hydrophilic pocket (Pi site or new Pi site) for inorganic Pi or the C3P of flopped GAP [48,49,50] (Fig. 4a, left magnified). Moreover, the strand-loop-helix covering residues 210–215 of region II undergoes conformational changes to form a more compact catalytic center [48, 49]. The decrease in the deuterium exchange rate in region II might be attributed to the displacement of inorganic phosphate (Pi) by PGG when interacting with GAPDH. The key residues interacting with NAD+ or Pi are protected from HDX upon PGG binding, strongly indicating a more stable interaction is formed between PGG and these regions, which is consistent with the competitive modality of PGG with respect to NAD+ and Pi in the kinetic assay.

a The GAPDH monomer was modeled with PGG to show two binding regions as revealed by HDX. The two regions are enlarged, and the critical amino acids are shown as sticks. b View of the tetramer interface regions encoded with HDX color. Regions III and IV show enhanced deuterium uptake upon binding to PGG, and key amino acids are shown as sticks.
Even though Ps is a part of the center pocket and PGG likely occupies the site, the critical amino acids (Thr182 and Arg234) of the Ps site did not show a decrease in deuterium uptake, indicating the possibility that PGG does not occupy the Ps site. We then performed in silico docking of PGG to this site and found that PGG fits very well within it, with a docking score ranging from −9.65 to −10.94 of top 10 ranking. The docking pose (Fig. 4a, middle) harboring the interactions formed between PGG to region I and II also vacated the Ps site, yielding a docking score of −9.86, ranking 5th. Accordingly, this GAPDH pocket is positively charged at pH 7.4, which effectively attracts the negatively charged PGG (Supplementary Fig. S5a).
Interestingly, both regions III and IV are located at the tetramerization interface of GAPDH (Fig. 4b, middle), especially region III, which has a conserved S-shaped loop (S loop; residues 180–206) that forms the tetrameric core. The increased deuterium incorporation of regions III and IV suggest enhanced flexibility of this region upon PGG binding. Since these regions are vital for GAPDH tetramer formation, it is possible that the enhanced flexibility of these regions upon PGG binding reduces the stability of the intact tetramer. Dynamic light scattering did not indicate significant depolymerization effects on the GAPDH tetramer (Supplementary Fig. S5b), suggesting that PGG may affect the local conformation, not disrupt the tetramer. Notably, regions III and IV are distant from the potential binding sites, e.g., regions I, II, and III, and the dynamic nature of the GAPDH tetramer enables its recognition with PGG and permits PGG-dependent allosteric regulation at distal regions. Region III covers many amino acids that not only interact with intramolecular NAD+ but also contact intersubunit NAD+ (Fig. 4b, right magnified). Region IV has no direct contact with NAD+ or GAP, but the flank helix interacts with the other two subunits (Fig. 4b, left magnified). Hence, PGG binding may directly regulate the interface region of GAPDH.
We thus speculated that PGG binds to region I and region II of GAPDH and possibly exerts its inhibitory effect by the following main mechanisms. PGG competes with NAD+ through a partially occupied NAD+ pocket and may form interactions with residues interacting with NAD+, such as Glu97 Ser98, Thr99, Gly100, and Phe102. In parallel, PGG may enable GADPH to adopt a loosened tetramer conformation due to enhanced flexibility at the interface. Hence, the impaired stability of the GAPDH tetramer may lead to partial inhibition of key downstream signals. In summary, the unique HDX-MS results combined with structure modeling data suggest that PGG may bind to the center pocket partially overlapping with NAD+ and Pi. Furthermore, PGG can affect not only NAD+ binding but also the stability of the GAPDH tetramer, suggesting that PGG has a previously unreported mechanism of action in GAPDH inhibition. Our results shed light on designing potential inhibitors occupying the pocket composed of region I, region II, and region III.
PGG directly engages in GAPDH cellular functions
To determine whether PGG engages in a cellular context, we evaluated PGG GAPDH inhibition activities in HCT116 cancer cells and RAW264.7 immune cells. Lysates of HCT116 and RAW264.7 cells were precipitated with saturated (NH4)2SO4 to remove endogenous NADH or NADPH, and the precipitate was resuspended in enzyme assay buffer to determine the IC50. PGG showed an IC50 of 8.4 ± 0.13 μM (Fig. 5a) in HCT116 cells and an IC50 of 16.8 ± 0.57 μM (Supplementary Fig. S6a) in RAW264.7 cells, indicating that PGG can indeed target cellular GAPDH. Next, to confirm that PGG potentiated the inhibition of GAPDH specific, small interfering RNA oligos were transfected into HCT116 cells to knockdown endogenous GAPDH. Western blot analysis with anti-GAPDH showed efficient knockdown of GAPDH (Supplementary Fig. S6b) in the HCT116 cells, which were called siHCT116 cells. SiHCT116 cells and siNC control HCT116 cells were also treated with PGG and then subjected to evaluation of cell lysate enzyme activity. As shown in Fig. 5b, the efficient knockdown of GAPDH in siHCT116 cells resulted in only 50% GAPDH activity in the control cells, implying intrinsic high enzymatic activity of GAPDH. PGG inhibited 80% of the GAPDH activity in siNC control cells and 20% of the GAPDH activity in knockdown cells at a concentration of 200 μM. Collectively, these data suggested that PGG is a cell-active inhibitor of GAPDH.

a PGG inhibitory activity of cellular GAPDH was determined in HCT116 cell lysate. The experiment was repeated three times, and error bars represent the SEM. b Relative GAPDH activity was determined in GAPDH-knockdown HCT116 cell lysate and siNC control-expressing HCT116 cell lysate. Each point represents three replicates, and the error bars represent the SEM. c Glucose consumption and (d) lactate production in RAW264.7 cells treated with or without PGG for 12 h. Basic DMEM containing 25 mM glucose, 2 mM glutamine, and 10% dialyzed fetal bovine serum (FBS) was used to culture RAW264.7 cells for this assay. The data represent three parallel experiments, and the error bars represent the standard error of the mean (SEM). *P < 0.05, **P < 0.01, ****P < 0.0001. Each group was compared by Student’s t test.
PGG inhibits macrophage activation and improves the survival rate of LPS-treated mice
Mechanistically, molecules that inhibit GAPDH also result in the downregulation of the cell glycolysis pathway, leading to decreased glucose consumption and lactate production. We examined the remaining glucose concentration and the lactate content in the supernatant medium of LPS-treated RAW264.7 cells in the presence or absence of the indicated concentration of PGG. Both glucose consumption and lactate production were significantly decreased in the 25 μM PGG-treated group compared to the DMSO-treated group (Fig. 5c, d). Studies have reported that macrophages challenged by endotoxin, i.e., lipopolysaccharide, shifts its metabolic state rapidly to glycolysis, and a sharp increase in the rate of glycolysis is necessary for macrophage activation [4, 51]. Therefore, we explored whether PGG can inhibit macrophage activation. The proinflammatory cytokines interleukin (IL)-1β and inducible nitric oxide synthase (iNOS) are almost always increased in classically activated macrophages (M1), which are usually considered signatures of M1 macrophages. In LPS-challenged RAW264.7 macrophages, PGG significantly decreased the mature form and transcription of IL-1β (Fig. 6a, c). Accordingly, the synthesis (Fig. 6b) and transcription (Fig. 6d) of iNOS were inhibited by PGG at 25 μM. Our data supported the idea that PGG can inhibit GAPDH and downregulate glycolysis to repress the activation of LPS-activated macrophages.

a, b RAW264.7 cells were pretreated with the indicated concentration of PGG for 2 h and then stimulated with 1 μg/mL LPS for 4 h. The cells were collected for detecting the mature forms of IL-1β and iNOS n by Western blotting. c, d RAW264.7 cells were treated the same as in (a and b). The cells were harvested for IL-1β mRNA and iNOS mRNA level evaluation by RT-qPCR. The data represent three parallel experiments, and the error bars represent the SEM. e RAW264.7 cells were pretreated with the indicated concentration of PGG for 2 h and then stimulated with 1 μg/mL LPS for 4 h. The cell supernatant was collected for TNFα secretion content determination by an ELISA kit, each data point represents a duplicate assay, and the mean ± SEM is shown. f RAW264.7 cells were treated the same as in (e). The cells were harvested for IL-6 mRNA evaluation by RT-qPCR. The data represent three parallel experiments, and the error bars represented the SEM. *P < 0.05, ***P < 0.001, ****P < 0.0001. Each group was compared by Student’s t test. g The survival rate of (LPS)/D-galactosamine (GalN)-challenged mice over with 60 mg/kg of PGG or vehicle control for 24 h. Each group included 10 mice. Student’s t test was used to compare the PGG treated groups to the vehicle-treated group, with the mice in all groups challenged by LPS, *P < 0.05.
Given the capacity of PGG to restrain macrophage activation and reduce the release of a series of proinflammatory cytokines (TNFα and IL-6) (Fig. 6e, f), we further investigated the anti-inflammatory effect of PGG in an LPS-/GalN-induced acute liver injury mouse model. We assessed whether PGG can improve the mouse survival rate of lipopolysaccharide (LPS)/D-galactosamine (GalN)-challenged mice over 24 h. As expected, the survival rate of the 60 mg/kg PGG treatment group (Fig. 6g) was improved significantly compared to that of the vehicle group. These data suggested that PGG inhibition of GAPDH can alleviate proinflammatory immune cell activation.
Discussion
Aerobic glycolysis, also known as the Warburg effect, is a hallmark of cancer cell glucose metabolism and plays a crucial role in the activation of various types of immune cells. Small molecules that inhibit the glycolytic pathway have been widely studied and advanced into clinical applications. Most notably, 2-deoxy-2-[fluorine-18] fluoro-D-glucose (18F-FDG), an analog of glucose, has been a valuable tool for cancer diagnosis, staging, restaging, and predicting therapy outcomes [52, 53]. Many efforts have been invested in the development of drugs targeting the rate-limiting enzymes (hexokinase, phosphofructokinase, and pyruvate kinase) of glycolysis. However, development of most of these enzymes have stagnated in clinical trials [23]. Accumulating evidence indicated that GAPDH plays a pivotal role in cancer cell proliferation and invasion and excessive immune cell activation [3, 5, 6, 23, 24, 51]. Furthermore, recent data suggest that dimethyl fumarate can directly inactivate GAPDH, revealing a new effective mechanism of this drug in multiple sclerosis treatment [22] and indicating that molecules targeting GAPDH have therapeutic potential. Thus, highly potent modulators selective for GAPDH may be powerful tools for GAPDH physiological and pathological function exploration. Here, we designed and developed an easy-to-handle and cost-effective high-throughput format assay for GAPDH inhibitor discovery. The assay is sufficiently robust for automatic or manual screening of diverse chemical libraries and can be adopted by other laboratories to screen their unique compound libraries. Through our assay, we discovered that the natural product PGG is a potent, reversible, and competitive inhibitor of GAPDH with Ki = 0.5 μM. We demonstrated that PGG can inhibit GAPDH enzymatic activity in vitro and in vivo; furthermore, PGG also downregulated the GAPDH-dependent glycolysis pathway in LPS-stimulated macrophages.
Molecules such as selegiline, KA, and DMF, have greatly promoted the advancement of the exploration of GAPDH therapeutic potential. These inhibitors function through common suicide inactivation by covalently binding to the catalytic cysteine residue of GAPDH. The kinetic assay indicated that PGG is a reversible inhibitor of GAPDH, indicating that PGG has a different mechanism of action than irreversible inhibitors. The steady-state kinetic assay demonstrated that PGG is competitive with NAD+ and inorganic phosphate. In-depth HDX-MS in a combination of in silico molecular docking revealed that PGG probably fits in the center pocket that is composed of the inorganic phosphate site, C3-phosphate (C3P) group of the GAP binding site (Ps site), and nicotinamide moiety of NAD+. In addition, PGG was the first molecule to bind to this pocket without covalently modifying the catalytic cysteine, indicating that molecules without a covalent moiety that can efficiently occupy this pocket can inhibit GAPDH activity. Notably, PGG does not fully occupy the center pocket, most likely because PGG overlaps the site of the nicotinamide moiety and inorganic phosphate. Overall, the unique binding site of PGG indicates that the center pocket is a new and easy-to-target pocket for which more potential and highly selective irreversible or reversible inhibitors of GAPDH can be designed.
PGG has five galloyl groups and seems to be a pan-assay interference compound. However, a series of experiments validated PGG as a true inhibitor of GAPDH. First, one-dimensional NMR spectroscopy and microscale thermophoresis analysis verified that PGG is not a nonspecific aggregate of GAPDH. Second, the dynamic scatter light assay confirmed that PGG does not allow GAPDH to aggregate. Third, kinetic assay data indicated that PGG competes with NAD+ and Pi, suggesting that PGG did not coat GAPDH that sequester substrates. Moreover, in the screening assay, PGG showed no absorption at 450 nm without GAPDH. Finally, the HDX-MS assay showed that PGG interacts with GAPDH in a specific region, not solely in random areas.
PGG, a galloyl-beta-D-glucose compound having five galloyl groups in the 1, 2, 3, 4, and 6 positions, is flexible in solution challenged the GAPDH-PGG complex crystal structure packing. We failed to obtain the GAPDH-PGG complex structure either as cocrystals or by soaking. Nonetheless, the data presented by HDX-MS can be used to map the critical amino acids involved in GAPDH and PGG interactions. Furthermore, the HDX-MS data showed that the tetramer interface of GAPDH has enhanced flexibility upon PGG binding, which may impair the stability of the tetramer. The data presented in this paper suggest that HDX-MS is an effective tool for investigating the interactions of proteins with small molecules. Moreover, HDX-MS can provide information about the dynamic conformation change in the presence and absence of small molecules, which is nearly impossible to discern with the static crystal structure. Thus, HDX-MS is a powerful tool to study the interaction of proteins and small molecules, particularly when the molecules bind to proteins resulting in significant conformation changes that cannot be sufficiently assessed by the crystal structure.
In summary, we developed a high-throughput assay to screen novel GAPDH inhibitors. Through this method, we discovered that the natural product PGG, which is enriched in many traditional herbs, is a reversible inhibitor of GAPDH. PGG inhibits GAPDH in vitro with high potency, Ki = 0.48 ± 0.01 μM. PGG reduced glucose consumption and lactate production in LPS-stimulated macrophages, decreased proinflammatory cytokine secretion, and improved the survival rate of an LPS/GalN-induced mouse model. In contrast to the well-known koningic acid and the approved drugs selegiline and dimethyl fumarate, which covalently modify catalytic cysteine 152 of GAPDH, PGG represents a new class of GAPDH inhibitors. Classic steady-state assays indicated that PGG is reversible and competes with the cofactor NAD+ and inorganic phosphate. HDX-MS demonstrated that PGG binds to fragments 96–102 and 206–218 to disrupt NAD+ and inorganic phosphate affinity for GAPDH, respectively. Importantly, two regions at the tetramer interface showed enhanced flexibility upon PGG binding, which may lead to an intact yet subtly conformationally altered tetramer. Thus, the center pocket of GAPDH is a targetable site of GAPDH. The unique mechanism of PGG action makes it as a great initial compound for developing additional potent and reversible GAPDH inhibitors.
References
Warburg O. The metabolism of carcinoma cells. J Cancer Res. 1925;9:148–63.
Warburg O. On the origin of cancer cells. Science. 1956;123:309–14.
Geltink RIK, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol. 2018;36:461–88.
Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–84.
Palsson-McDermott EM, O’Neill LAJ. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020;30:300–14.
Patel CH, Leone RD, Horton MR, Powell JD. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug Discov. 2019;18:669–88.
Seth Nanda C, Venkateswaran SV, Patani N, Yuneva M. Defining a metabolic landscape of tumours: genome meets metabolism. Br J Cancer. 2020;122:136–49.
Martin WF, Cerff R. Physiology, phylogeny, early evolution, and GAPDH. Protoplasma. 2017;254:1823–34.
Murthi P, Fitzpatrick E, Borg AJ, Donath S, Brennecke SP, Kalionis B. GAPDH, 18S rRNA and YWHAZ are suitable endogenous reference genes for relative gene expression studies in placental tissues from human idiopathic fetal growth restriction. Placenta. 2008;29:798–801.
Chauhan AS, Kumar M, Chaudhary S, Patidar A, Dhiman A, Sheokand N, et al. Moonlighting glycolytic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH): an evolutionarily conserved plasminogen receptor on mammalian cells. FASEB J. 2017;31:2638–48.
Guha P, Harraz MM, Snyder SH. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc Natl Acad Sci USA. 2016;113:1417–22.
Chuang DM, Ishitani R. A role for GAPDH in apoptosis and neurodegeneration. Nat Med. 1996;2:609–10.
Waingeh VF, Lowe SL, Thomasson KA. Brownian dynamics of interactions between glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mutants and F-actin. Biopolymers. 2004;73:533–41.
Kosova AA, Khodyreva SN, Lavrik OI. Role of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in DNA Repair. Biochem (Mosc). 2017;82:643–54.
Nicholls C, Pinto AR, Li H, Li L, Wang L, Simpson R, et al. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) induces cancer cell senescence by interacting with telomerase RNA component. Proc Natl Acad Sci USA. 2012;109:13308–13.
Hara MR, Thomas B, Cascio MB, Bae BI, Hester LD, Dawson VL, et al. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci USA. 2006;103:3887–9.
Olah J, Tokesi N, Vincze O, Horvath I, Lehotzky A, Erdei A, et al. Interaction of TPPP/p25 protein with glyceraldehyde-3-phosphate dehydrogenase and their co-localization in Lewy bodies. FEBS Lett. 2006;580:5807–14.
Allen M, Cox C, Belbin O, Ma L, Bisceglio GD, Wilcox SL, et al. Association and heterogeneity at the GAPDH locus in Alzheimer’s disease. Neurobiol Aging. 2012;33:203 e25–33.
Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, et al. A predictive model for selective targeting of the warburg effect through GAPDH inhibition with a natural product. Cell Metab. 2017;26:648–59 e8.
Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–51.
Galvan-Pena S, Carroll RG, Newman C, Hinchy EC, Palsson-McDermott E, Robinson EK, et al. Malonylation of GAPDH is an inflammatory signal in macrophages. Nat Commun. 2019;10:338.
Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. 2018;360:449–53.
Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635–49.
O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–65.
Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. 2019;16:425–41.
Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–74.
Birkmayer W, Riederer P, Youdim MB, Linauer W. The potentiation of the anti akinetic effect after L-dopa treatment by an inhibitor of MAO-B, Deprenil. J Neural Transm. 1975;36:303–26.
Kragten E, Lalande I, Zimmermann K, Roggo S, Schindler P, Muller D, et al. Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(-)-deprenyl. J Biol Chem. 1998;273:5821–8.
Unni S, Deshmukh P, Krishnappa G, Kommu P, Padmanabhan B. Structural insights into the multiple binding modes of Dimethyl Fumarate (DMF) and its analogs to the Kelch domain of Keap1. FEBS J 2021; 288: 1599–613.
Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369:1633–7.
Cannell RJP, Farmer P, Walker JM. Purification and characterization of pentagalloylglucose, an alpha-glucosidase inhibitor antibiotic from the fresh-water green-alga spirogyra-varians. Biochem J. 1988;255:937–41.
Adachi H, Konishi K, Kita K, Horikoshi I. Inhibitory effects of pentagalloylglucose on the respiratory-chain of photobacterium-phosphoreum. Chem Pharm Bull. 1988;36:2499–505.
Briggs G, Haldane JJTBj. A note on the kinetics of enzyme action. Biochem J. 1925;19:338–9.
Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, et al. HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spectrom. 2012;23:1512–21.
Wolfson-Stofko B, Hadi T, Blanchard JS. Kinetic and mechanistic characterization of the glyceraldehyde 3-phosphate dehydrogenase from Mycobacterium tuberculosis. Arch Biochem Biophysics. 2013;540:53–61.
Ishiyama M, Shiga M, Sasamoto K, Mizoguchi M, He P-g. A new sulfonated tetrazolium salt that produces a highly water-soluble formazan dye. Chem Pharm Bull. 1993;41:1118–22.
Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73.
Barnes EC, Kumar R, Davis RA. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat Prod Rep. 2016;33:372–81.
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134:17963–71.
McClary B, Zinshteyn B, Meyer M, Jouanneau M, Pellegrino S, Yusupova G, et al. Inhibition of eukaryotic translation by the antitumor natural product agelastatin A. Cell Chem Biol. 2017;24:605-+.
Cui ZM, Liu Y, Wan W, Xu YY, Hu YH, Ding M, et al. Ethacrynic acid targets GSTM1 to ameliorate obesity by promoting browning of white adipocytes. Protein Cell. 2020. https://doi.org/10.1007/s13238-020-00717-7.
Wang Z, Li ZX, Zhao WC, Huang HB, Wang JQ, Zhang H, et al. Identification and characterization of isocitrate dehydrogenase 1 (IDH1) as a functional target of marine natural product grincamycin B. Acta Pharmacol Sin. 2020. https://doi.org/10.1038/s41401-020-0491-6.
Feldman KS, Sahasrabudhe K, Lawlor MD, Wilson SL, Lang CH, Scheuchenzuber WJ. In vitro and in vivo inhibition of LPS-stimulated tumor necrosis factor-alpha secretion by the gallotannin beta-D-pentagalloylglucose. Bioorg Med Chem Lett. 2001;11:1813–5.
Zhang JH, Li L, Kim SH, Hagerman AE, Lu JX. Anti-cancer, anti-diabetic and other pharmacologic and biological activities of penta-galloyl-glucose. Pharm Res. 2009;26:2066–80.
Li FJ, Liu Y, Yuan Y, Yang B, Liu ZM, Huang LQ. Molecular interaction studies of acetylcholinesterase with potential acetylcholinesterase inhibitors from the root of Rhodiola crenulata using molecular docking and isothermal titration calorimetry methods. Int J Biol Macromol. 2017;104:527–32.
Chalmers MJ, Busby SA, Pascal BD, He YJ, Hendrickson CL, Marshall AG, et al. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2006;78:1005–14.
Wei H, Mo JJ, Tao L, Russell RJ, Tymiak AA, Chen GD, et al. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discov Today. 2014;19:95–102.
Yun M, Park CG, Kim JY, Park HW. Structural analysis of glyceraldehyde 3-phosphate dehydrogenase from Escherichia coli: direct evidence of substrate binding and cofactor-induced conformational changes. Biochemistry. 2000;39:10702–10.
Moniot S, Bruno S, Vonrhein C, Didierjean C, Boschi-Muller S, Vas M, et al. Trapping of the thioacylglyceraldehyde-3-phosphate dehydrogenase intermediate from Bacillus stearothermophilus — direct evidence for a flip-flop mechanism. J Biol Chem. 2008;283:21693–702.
Chaikuad A, Shafqat N, Al-Mokhtar R, Cameron G, Clarke AR, Brady RL, et al. Structure and kinetic characterization of human sperm-specific glyceraldehyde-3-phosphate dehydrogenase, GAPDS. Biochem J. 2011;435:401–9.
Jung J, Zeng H, Horng T. Metabolism as a guiding force for immunity. Nat Cell Biol. 2019;21:85–93.
Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res. 2005;11:2785–808.
Almuhaideb A, Papathanasiou N, Bomanji J. 18F-FDG PET/CT imaging in oncology. Ann Saudi Med. 2011;31:3–13.
Acknowledgements
We are grateful to the National Centre for Protein Science Shanghai (Protein Expression and Purification system) for their instrument support and technical assistance. We gratefully acknowledge the financial support from the National Natural Science Foundation of China (81728020 to KHZ, 91853205, 81625022, and 81821005 to CL, 21820102008 to HLJ), K. C. Wong Education to CL, the Science and Technology Commission of Shanghai Municipality (19XD1404700 to CL and 18431907100 to HLJ), the Chinese Academy of Sciences (CASIMM0120184015 to CL). KHZ is supported by Taishan Scholar. Part of the study is funded by the Top Talents Program for One Case Discussion of Shandong Province.
Author information
Authors and Affiliations
Contributions
WL, LPL, KHZ and CL conceived the project, provided intellectual contributions, designed experiments, performed and analyzed experiments, and wrote the paper. NS and JZ performed the HDX-MS experiment and described the method used to perform this experiment. YLD and NXZ performed the NMR experiment and described the method used to perform the NMR experiment. YJL provided advice for the animal experiment. CPL provided expert advice on enzymology. YYZ, ZYS, XRZ, JL, HBW, and SHX provided help with the protein expression and high-throughput assays. KXC and HLJ read the paper and provided valuable suggestions about the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Li, W., Liao, Lp., Song, N. et al. Natural product 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose is a reversible inhibitor of glyceraldehyde 3-phosphate dehydrogenase. Acta Pharmacol Sin 43, 470–482 (2022). https://doi.org/10.1038/s41401-021-00653-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00653-0
