
Abstract
Nuclear receptors (NRs) are members of a large superfamily of evolutionarily related transcription factors that control a plethora of biological processes. NRs orchestrate complex events such as development, organ homeostasis, metabolism, immune function, and reproduction. Approximately one-half of the 48 human NRs have been shown to act as ligand-regulated transcription factors and respond directly to a large variety of endogenous hormones and metabolites that are generally hydrophobic and small in size (eg, retinoic acid or estradiol). The second half of the NR family comprises the so-called orphan receptors, for which regulatory ligands are still unknown or may not exist despite the presence of a C-terminal ligand-binding domain, which is the hallmark of all NRs. Several chemicals released into the environment (eg, bisphenols, phthalates, parabens, etc) share some physicochemical properties with natural ligands, allowing them to bind to NRs and activate or inhibit their action. Collectively referred to as endocrine disruptors or endocrine-disrupting chemicals (EDCs), these environmental pollutants are highly suspected to cause a wide range of developmental, reproductive, neurological, or metabolic defects in humans and wildlife. Crystallographic studies are revealing unanticipated mechanisms by which chemically diverse EDCs interact with the ligand-binding domain of NRs. These studies thereby provide a rational basis for designing novel chemicals with lower impacts on human and animal health. In this review, we provide a structural and mechanistic view of endocrine disrupting action using estrogen receptors α and β, (ERα/β), peroxisome proliferator activated receptor γ (PPARγ), and their respective environmental ligands as representative examples.
Similar content being viewed by others
Introduction
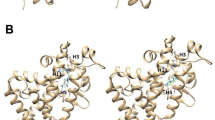
The combination of a large number of molecular, structural, and genetic studies has provided a comprehensive view on the actions of nuclear receptors (NRs) — from the basic molecular events to the effects of receptor ablation on animal development and physiology1,2. NRs are a family of transcription factors that regulate cognate gene networks, resulting in profound physiological changes. Consequently, dysfunctional NR signaling leads to proliferative, reproductive, and metabolic diseases, such as cancer, infertility, obesity, or diabetes. NR-based pharmaceuticals are among the most commonly used drugs. The 48 NRs are modular proteins organized into three major functional domains, namely (i) a variable and intrinsically unfolded N-terminal A/B domain harboring the transcriptional activation function 1 (AF-1), (ii) a conserved DNA-binding domain (DBD), and (iii) a C-terminal ligand-binding domain (LBD) hosting the activation function 2 (AF-2)3. The crystal structures of several NR LBDs have been determined, revealing a conserved core of 12 α-helices and a short two-stranded antiparallel β-sheet arranged into a three-layered sandwich fold generating a predominantly hydrophobic cavity that may accommodate ligands. Ligand binding relies on the intrinsic dynamics of the small molecule and of the ligand-binding pocket (LBP) that allows a mutual adaptation of both partners4,5,6. The structures also showed that depending on the nature of the bound ligand, this domain can adopt two predominant conformations, which differ primarily in the position of the C-terminal helix H127 (Figure 1). Upon binding of an agonist (the natural ligand or a synthetic compound mimicking its action), H12 adopts a stable active position, which allows the receptor to interact with transcriptional coactivators. The interaction surface of the receptor is built up from amino acids residing in the helices H3, H4, and H12, which form a predominantly hydrophobic groove designed to recognize small LxxLL helical motifs present in coactivators. By contrast, binding of an antagonist (generally obtained by chemical synthesis) prevents the positioning of H12 in the active conformation and redirects the helix to the coactivator binding site, where it adopts an inactive conformation and precludes coactivator interaction. Further biophysical analyses revealed that partial agonists/antagonists correspond to a third class of NR ligands that fail to stabilize either of the aforementioned conformations8. In the presence of partial agonists/antagonists, the conformational dynamics of H12 remains high; therefore, the activity profile of these compounds largely depends on the relative abundance of coactivators and corepressors in a cell. They act as cell-selective modulators with agonistic or antagonistic properties according to the cellular context. The fourth class of NR ligands gathers compounds that stabilize the interaction with corepressors and are thus referred to as inverse agonists9,10,11. Thus, NRs should be viewed as molecular dimmers whose activity can be finely tuned by natural, pharmaceutical, or environmental ligands displaying specific chemical features. It is worth noting that while NR ligands regulate AF-2 activity through direct binding to LBDs and allosteric conformational changes, they also modulate AF-1 activity through domain-domain (Nt–Ct) interactions.

The position of helix H12 determines receptor activity. Upon binding of an agonist molecule, H12 adopts a stable active position, which allows the receptor to interact with transcriptional coactivators (CoA) (active form, bottom left). Binding of an antagonist ligand redirects H12 to the coactivator binding site, precluding coactivator interaction (inactive form, bottom middle). Inverse agonist compounds stabilize the interaction with corepressors (CoR) (repressive form, bottom right). Molecular representations were generated using the PyMOL software (http://www.pymol.org/).
Numerous synthetic substances released into the environment through human activities have been shown to mimic or interfere with the action of endogenous hormones and act as endocrine-disrupting chemicals (EDCs), causing reproductive, developmental, metabolic, or neurological diseases, as well as hormone-related cancers12,13,14. Several EDCs are man-made compounds, such as bisphenols (BPs), phthalates, parabens, dioxins, pesticides, alkylphenols, organotins, polychlorinated biphenyls, and perfluoroalkyl compounds. Additionally, some natural EDCs can be found in plants and fungi. For more than 20 years, laboratory animals and epidemiological studies have highlighted the pivotal role of NRs in transducing several adverse effects of these chemicals. In addition to the bona fide endogenous ligands, the predominantly hydrophobic pocket enclosed in the LBD of NRs can accommodate small lipophilic exogenous compounds with affinities ranging from sub-nanomolar to micromolar values. Certain compounds, such as bisphenol-A (BPA), benzyl-butyl-phthalate (BBP), 4-tert-octylphenol (4-OP), butylparaben (BPB), or chlordecone (CLD), have been reported to activate the estrogen receptors (ERα and β) and act as antagonistic ligands of the androgen receptor (AR). The xenobiotic receptor PXR (pregnane X receptor) is the target of numerous environmental compounds and natural molecules. However, the affinity of this receptor for chemicals is generally relatively low (micromolar range). The peroxisome proliferator activated receptor γ (PPARγ) binds to and is activated by molecules such as brominated or chlorinated derivatives of BPA (TBBPA, TCBPA), perfluorinated compounds (PFOS and PFOA), and certain phthalates, such as the mono-ethylhexyl phthalate (MEHP). Several other NRs are responsive to environmental compounds, including the progesterone (PR), glucocorticoid (GR), mineralocorticoid (MR), thyroid hormone (TRα and β), constitutive androstane (CAR), and retinoid X (RXRα, β, and γ) receptors, as well as the peroxisome proliferator activated receptor α (PPARα) or the estrogen-related receptor γ (ERRγ)15. Considering the conservation of the LBD throughout the NR family and the huge chemical diversity of environmental compounds, all NR family members, including the orphan receptors, represent potential targets of environmental contaminants.
We and other groups have demonstrated the importance of structural studies to reveal the molecular details of the interaction between NRs and compounds structurally and chemically divergent from natural ligands15. They show that environmental pollutants bind to NRs via diverse sets of protein-ligand interactions, thus reflecting their differential activities, binding affinities, and specificities. In this review, a detailed analysis of the various binding/activation mechanisms will be presented using ERα/β and PPARγ as representative examples. As described below, a large set of environmental ligands has been identified for these NRs.
The estrogen receptors and their environmental ligands
The estrogen receptors in health and disease
Estrogen signaling is primarily mediated by the two estrogen receptors: ERα (also called NR3A1) and ERβ (also called NR3A2)16,17. Similar to most NRs, ERs bind as dimers to DNA response elements in the promoter region of the target genes and respond to the naturally occurring sex hormone 17β-estradiol (E2; Table 1) and its metabolites, estrone and estriol, through interaction with coregulator complexes and regulation of target gene expression. The amino acid sequence identity between ERα and ERβ is approximately 97% in the DBD and approximately 56% in the LBD, whereas the N-terminus is poorly homologous at 24%. Transcriptional activation by ERs is mediated by the two distinct activation functions AF-1 and AF-2, the relative importance of which depends on cellular and promoter contexts. Both ERs are widely expressed throughout the body, but present different tissue distributions and functions18,19. ERα is primarily expressed in the uterus, liver, kidney, and heart, whereas ERβ is expressed primarily in the ovary, prostate, lung, gastrointestinal tract, bladder, and hematopoietic and central nervous systems. However, ERα and ERβ are co-expressed in a number of tissues, including the mammary, thyroid, and adrenal glands, the bone, and some regions of the brain. Although ERα and ERβ share similar mechanisms of action, several differences in the transcriptional abilities of each receptor and distinct phenotypes between gene-null animals have been identified, suggesting that these receptors may regulate distinct cellular pathways. The respective functional roles of ERα and ERβ in physiology and disease might result from a complex interplay between the expression levels of each ER, the relative affinity for a specific response element, ligand and cofactor availability, and interaction with other transcription factors. Additionally, ERs can be activated through post-translational modifications and can perform non-genomic signaling20.
The primary function of estrogens and their receptors is to regulate female reproduction. They play key roles in the growth and maintenance of the mammary gland and the uterus, promote the formation of female secondary sex characteristics, regulate reproductive cycles, and affect sexual and maternal behavior. In addition, ERs have pleiotropic regulatory roles in a diverse range of tissues, such as the cardiovascular or central nervous systems, bone, prostate, and adipose tissue. They are involved in the regulation of bone density, blood lipid levels, fat deposition, and brain functions19. The importance of ERs in physiology is evidenced in menopausal women, whose endogenous estrogen levels decrease, which then increases the risk of osteoporosis, cardiovascular diseases, and brain disorders (Alzheimer's and Parkinson's diseases). Furthermore, in addition to controlling the normal development and function of the reproductive system and other tissues, estrogens are key regulators of primary breast and prostatic cancer growth17. Approximately 40% of human cancers require steroid hormones for their growth, and the first-line of treatment for hormone-dependent cancers is based on androgen and estrogen antagonists that interact with AR or ERs and shut down the corresponding hormone-responsive pathway. ERβ has been shown to antagonize the effects mediated by ERα on cell proliferation in the breast, uterus, ovary, and prostate21,22,23. In this regard, estrogens with selectivity for either ER subtypes may produce different biological outcomes, particularly on cancer cell proliferation. Considering the widespread role of ERs in human physiology, it is not surprising that environmental compounds that bind to ERs (thereby substituting for the endogenous hormone and deregulating the finely tuned action of E2) can lead to ER-related disorders. These include breast, endometrial, colorectal, and prostate cancers, as well as neurodegenerative, inflammatory, immune, cardiovascular, and metabolic diseases. In this review, we describe the mechanisms by which compounds that are structurally divergent from natural estrogens and that belong to families representing pollutants bind to ERs and impact their signaling pathways.
Environmental estrogens
A large group of structurally diverse compounds with different binding modes
Cell-based assays and laboratory studies on animals have revealed that the group of molecules acting as ER environmental ligands (xenoestrogens) is highly heterogeneous and includes both natural and synthetic compounds (Table 1). Naturally occurring xenoestrogens, such as phyto-estrogens (eg, genistein and ferutinine) and myco-estrogens (eg, zearalenone) are found in plants and fungi, whereas synthetic estrogens comprise both pharmaceuticals, such as ethinylestradiol (EE2; an active component of contraceptive pills) or diethylstilbestrol (DES; used until the 1970s to prevent miscarriage in women with high risk pregnancies) and a variety of industrial chemicals, such as alkylphenols, bisphenols, and their halogenated derivatives, parabens, phthalates, or benzophenones. The pharmaceuticals and some natural estrogens are the ligands that bind to ERs with the highest affinity, with dissociation constant (Kd) values in the (sub)nanomolar range24. For example, DES and α-zearalanol, a metabolite of zearalanone, display an affinity of 0.1–0.4 nM for ERs. Industrial compounds, by contrast, exhibit a significantly lower affinity for ERs, with Kd values ranging from 10 nM to 10 μM24. Representative examples are the plasticizers bisphenol-C (BPC; 20–40 nM) and bisphenol-A (BPA; 0.5–1.0 μM), the UV filter benzophenone-2 (BP-2; 0.1–0.5 μM), and the pesticide chlordecone (CLD; ∼5.0 μM). Transactivation and binding assays demonstrated that most xenoestrogens bind to both ER subtypes with similar affinities; however, genistein (GEN) binds slightly more strongly to ERβ than ERα25,26.
Crystallographic studies have revealed that in the ERs, the hormone binding pockets are lined with ∼18 hydrophobic residues that interact with the steroid scaffold of E2. Three polar residues located at the two ends of the pockets form hydrogen bonds with the phenolic and hydroxyl groups at the 3- and 17-positions of estrogen (Table 1). These polar residues are His524 (human ERα numbering) in helix H11 on one side and Glu353 (H3) and Arg394 (H5) on the other side (Figure 2A). Thus, the affinity and selectivity of hormone binding derives from both shape of the hydrophobic portion of the pocket and the presence of receptor-specific hydrogen-bond networks. Xenoestrogens exhibit a large structural diversity and vary both in size and chemical features. Several compounds contain two phenolic rings that mimic the A- and D-rings of E2 (Table 1). Accordingly, these ligands adopt a binding mode reminiscent of that used by E2, with the two phenol groups hydrogen-bonded to the polar residues of the LBP, as illustrated by BP-224 (Figure 2B) and the phytoestrogen resveratrol27 (RES, Figure 2C). The remaining contacts essentially involve van der Waals interactions, the number of which varies from one compound to another and accounts for the variation in binding affinities of the ligands. Another group of estrogenic ligands do not interact with His524 because they either lack a second hydroxyl group, such as ferutinine (FER), the surfactant 4-OP, dichlorodiphenyl-dichloroethylene (DDE), a metabolite of the pesticide DDT, and the preservatives butyl (BPB)- and propyl (PPB)-parabens (Figure 2D), or because they adopt a position that draws this hydroxyl moiety toward Thr347 in H3, such as BPC and bis-hydroxyphenyl-trichloroethane (HPTE), a metabolite of the pesticide methoxychlor (Figure 2E). Lastly, some chemicals, such as CLD or benzyl-butyl-phthalate (BBP), have no hydroxyl groups and display weak chemical proximity with E2. They are not engaged in any direct interaction with either of the polar residues (Figure 2F).

Xenoestrogens use diverse binding modes. Close-up views of the ligand-binding pocket of ERα in complex with estradiol (A), benzophenone-2 (B), resveratrol (C), propylparaben (D), HPTE (E), and benzyl-butyl-phthalate (F). Ligands and side chains of residues interacting with them are displayed in stick representation. The three key interacting residues Glu353, Arg394, and His524 are in blue, hydrophobic residues in light orange, and Thr347 in magenta. Oxygen, nitrogen, and chlorine atoms are displayed in red, blue, and green, respectively, and hydrogen bonds are indicated with dashed lines.
A wide array of activities associated with ERα and ERβ
In contrast to binding affinities, the activity of xenoestrogens depends on the receptor subtype and varies drastically among molecules that range from full agonists to weak agonists/antagonists. When bound to NR LBDs, an agonist (eg, the natural hormone E2) is involved in a multitude of interactions that stabilize key residue conformations, which are required to hold the activation helix H12 in the active position (Figure 1). Compounds that are engaged in sub-optimal interactions with surrounding residues and fail to stabilize the correct conformers will act as partial agonists or antagonists. This is the case for most ER environmental ligands: they are structurally diverse and often lack several chemical features of E2. Only a few of them mimic the natural hormone through well-conserved protein-ligand contacts.
BPs form a large family of chemicals that are commonly used in the manufacture of numerous consumer products. The most widely used BP is BPA (>3 million tons/year), which is found in plastics, food can linings, dentistry sealants, and thermal paper. Several other BPs are used in a variety of industrial applications, eg, BPC (Table 1) is used in the production of fire-resistant polymers. Many studies have revealed large contamination in the general population through food, drinking water, and skin absorption, with a direct correlation between BP exposure and the occurrence of reproductive, developmental, and metabolic diseases (obesity and diabetes) being highly suspected28. Cell-based assays revealed that BPA partially activates luciferase expression in MCF-7 and HeLa cells (inducing 60%–80% of the transactivation seen with E2) and triggers cell proliferation in a dose-dependent manner, albeit at a concentration higher than that of E229. The crystal structure of BPA-bound ERα LBD exhibits the active form of the receptor with H12 capping the LBP. BPA adopts a binding mode reminiscent of that used by E2 with the two phenol rings hydrogen-bonded to His524, Glu353, and Arg394 (Figure 3A)29. The remaining contacts involve 42 van der Waals interactions. Compared to the 51 contacts in the E2-containing structure, the smaller number of interactions accounts for the weaker affinity of BPA. The structure also reveals that key stabilizing interactions are missing in the ER-BPA complex, which possibly renders H12 more dynamic and accounts for the partial agonistic character of BPA. These missing or unfavorable contacts primarily involve H11 residues, including Gly521, His524, and Leu525 (Figure 3A). Because of their bulky chlorine atom substituents, the two bisphenols BPC and the closely related HPTE (Table 1) adopt a different binding mode compared to BPA24,29. A rotation of 180° around the main axis of phenol A-ring orients ring B toward H12 forming a new hydrogen bond with Thr347 in H3 (Figure 3B). In agonist-bound structures, this residue is involved in a network of van der Waals interactions. Thr347, Leu525, and Leu536 hold together H3, H11, and H12, three main components of the AF-2 function (Figure 3A). The 180° rotation of Thr347 in the BPC and HPTE complexes disrupts the stabilizing hydrophobic cluster. This is in agreement with the AF-2 antagonistic profiles of these compounds and their observed antagonist conformation, with H12 occupying the coactivator binding groove.

Structural determinants of xenoestrogens activity. Interaction network of bisphenol-A (A, in green) and bisphenol-C (B, in pink) with residues of the ERα ligand-binding pocket compared to that of estradiol (in teal), showing the agonist and antagonist position of bisphenols. Structure superposition of E2-bound ERβ LBD (in teal) with ferutinine-bound ERα LBD (C, in orange) or chlordecone-bound ERα LBD (D, in yellow). The presence of I373 in ERβ instead of M421 in ERα will induce a shift of bulky ligands towards helix H12, thus lowering the stability of AF-2. Oxygen, nitrogen, sulfur, and chlorine atoms are shown in red, blue, yellow, and green, respectively, and the interactions are indicated with dashed lines.
FER is a sesquiterpenoid found in the root of plants belonging to the genus Ferula, such as Ferula hermonis, native to the Middle Eastern region. The roots of F. hermonis have long been used in traditional medicine to treat menopausal disturbances in women. FER has an interesting activity profile that illustrates the subtype-dependence of xenoestrogen action. FER is a partial agonist of ERα but an ERβ antagonist30. Sequence and 3D-structure alignments reveal that the LBDs of ERα and ERβ share a high degree of homology in their primary sequence but exhibit two residue substitutions in their hormone binding pockets. These substitutions correspond to the replacement of Leu384 (ERα) by Met336 (ERβ) in H5 and Met421 (ERα) by Ile373 (ERβ) in H7. They account for most of the subtype-specific action of ER ligands31. Inspection of the crystal structures of both ER subtypes in complex with E2 reveals that the variable amino acids reside on each side of the C and D rings of E2 and create different space constraints in this portion of ERα and ERβ LBPs. The structure of FER in complex with ERα reveals that the ligand contains a bulky group that projects toward Met421, which in turn undergoes a large conformational change (Figure 3C). In ERβ, the branched and less flexible residue Ile373 is unable to move away from the pocket to make room for the ligand. Instead, a large shift of FER toward H12 possibly destabilizes the active conformation of H12, in line with the antagonistic activity of this compound in ERβ (Figure 3C). Interestingly, in contrast with ERα Met421 (H7), ERβ Met336 (H5) does not confer adaptability to the LBP of this receptor subtype, because strong constraints imposed by the surrounding residues maintain this residue in a single conformation24. Thus, with two bulky and rigid residues, ERβ appears more sensitive than ERα to ligand bulkiness and consequently, more easily antagonized by environmental compounds. Despite a fundamentally different chemical structure (Table 1), the organochlorine pesticide CLD displays a similar activity profile, acting as a weak ERα agonist and an ERβ antagonist32,33. This compound has been used as an agricultural pesticide to control the banana root borer from 1973 to 1993, particularly in the French West Indies. CLD undergoes very slow degradation in the environment, resulting in polluted soils and waterways, and remains a major source of human contamination through the consumption of locally produced foodstuffs. Recent studies have demonstrated that CLD accumulates in the body and that exposure at environmentally relevant levels is associated with a significant increase in the risk of prostate cancer, a decreased length of gestation, and negative effects on the cognitive and motor development of children34,35,36. Figure 3D shows that CLD occupies a smaller portion of the LBP than E2. However, it binds between ERα Met421 and Leu384, the LBP region that is sterically constrained in ERβ. With its cubic core and additional chlorine atoms, CLD displays a larger volume than the C- and D-rings of E2. Therefore, the binding mode of CLD in ERβ possibly interferes with the positioning of H12 in the active conformation. Because ERβ counteracts the action of ERα by exerting anti-proliferative and anti-inflammatory effects, the health impact of compounds acting as ERα agonists and ERβ antagonists (eg, FER, CLD, or BBP) is certainly higher than that of compounds with different subtype preferences. These compounds are highly associated with cancer incidence and tumor growth.
The peroxisome proliferator-activated receptor γ and its environmental ligands
The peroxisome proliferator-activated receptor γ in health and disease
The NR subfamily of peroxisome proliferator-activated receptors (PPARs) includes three members, PPARα (also called NR1C1), PPARβ/δ (also called NR1C2), and PPARγ (also called NR1C3). These receptors bind to PPAR-responsive DNA regulatory elements in the form of heterodimers with retinoid X receptor (RXR). They control the expression of genes involved in adipogenesis, glucose, lipid, and cholesterol metabolism37,38. PPARs bind and respond to dietary fatty acids and diverse lipid metabolites, including prostaglandins, eicosanoids37,39, and oxidized phospholipids. These receptors have different tissue distribution and physiological roles40. PPARα is expressed predominantly in the liver, heart, and brown adipose tissue, whereas PPARβ/δ is ubiquitously expressed. Both play a major role as activators of fatty acid oxidation pathways and therefore in the regulation of energy homeostasis. In addition, PPARα stimulates heme synthesis, cholesterol catabolism, and participates in the control of amino acid metabolism and urea synthesis. PPARβ/δ has a role in the control of cell proliferation and differentiation and is necessary for placental and gut development. PPARγ is highly expressed in adipose tissues and plays key roles in regulating adipogenesis41, lipid metabolism, and glucose homeostasis through improving insulin sensitivity42. Additionally, PPARγ is required for the function and survival of mature adipocytes. Lastly, several studies suggest that all three PPARs have anti-inflammatory activities38. However, the identification of specific endogenous PPARγ ligands has been difficult. In contrast with classical NRs, such as ERs that essentially bind and respond to a single specific ligand, it appears that natural PPARγ ligands are diverse and include unsaturated fatty acids, 15-deoxy-Δ12,14-prostaglandin J2, 9-hydroxy-octadecadienoic acid (Table 2), 15-hydroxy-eicosatetraenoic acid, prostaglandin J2, and nitrated fatty acids43,44.
Consistent with their tissue distribution and their role as sensors of lipids/fatty acids levels, regulating fatty acid catabolism, and lipid storage, all three PPARs are strongly implicated in the metabolic syndrome. Consequently, several PPAR subtype-specific synthetic ligands have been developed as drugs for treating metabolic diseases, such as hyperlipidemia or diabetes45. As PPARα agonists, fibrates, such as fenofibrate or clofibrate, are widely used to treat hypertriglyceridemia. By contrast, PPARγ is the target for antidiabetic agents of the thiazolidinedione (TZD) class, which includes troglitazone, pioglitazone, and rosiglitazone (full agonist reference molecule, Table 2). Because these compounds are potent activators of PPARγ, they exhibit not only robust insulin-sensitizing activities but also side effects associated with chronic activation, such as fluid retention, higher risk of cardiovascular disease, and weight gain42. However, a recent study has shown that classical agonism is not required for the antidiabetic effect of PPARγ ligands. Instead, the mechanism involves blockade of the phosphorylation of PPARγ by cyclin-dependent kinase 5 (Cdk5) at serine 27346. Novel antidiabetic compounds that have a unique mode of binding have been described in various reports47,48. They block the Cdk5-mediated phosphorylation without activating the transcriptional function of PPARγ and are associated with fewer side effects. Considering the physiological role of PPARγ in adipose tissue development and maintenance, it has been proposed that disruption of the regulatory pathways under the control of PPARγ may be involved in the onset of diabetes and obesity49. Activation of this receptor by certain xenobiotic compounds has been shown to stimulate adipogenesis in vitro and in vivo by inducing the differentiation of preadipocytes of the fibroblastic lineage into mature adipocytes50,51,52,53. This has led to the “obesogen hypothesis”, which states that in addition to the imbalance between caloric intake and expenditure, the rapidly growing obesity epidemic could implicate environmental risk factors, such as an increased exposure to chemicals that interfere with any aspects of metabolism52,54. Accordingly, compounds that have the potential to disrupt any metabolic signaling pathways and lead to increased fat accumulation and obesity are referred to as “obesogens”55. Below, we present illustrative examples of the group of environmental obesogens acting through the activation of PPARγ.
Environmental ligands of PPARγ
In the early 1990s, the study of a group of xenobiotic compounds termed peroxisome proliferators, which trigger hepatic and renal peroxisome proliferation in rodent cells, had led to the discovery of PPARs as a novel subfamily of NRs56. This set of compounds included phthalates, plasticizers, certain herbicides, and the fibrate class of hypolipidemic drugs. This group of PPAR disruptors now contains additional members, such as the biocides organotins, the perfluorooctanoic (PFOA) and perfluorooctanesulfonic (PFOS) acids, pharmaceuticals (eg, the PR antagonist RU-486), halogenated derivatives of BPA, the imidazole fungicide triflumizole, and some naturally occurring compounds, such as the polyphenols resveratrol, luteolin, and magnolol, which are found in plants. As shown in Table 2, the group of PPARγ environmental ligands is heterogeneous and contains a structurally and chemically disparate ensemble of molecules with very few shared molecular features, suggesting that each of them possibly interacts with the receptor through a specific mechanism.
PPARγ has a large Y-shaped ligand-binding pocket (LBP) with a volume of approximately 1440 Å3. The diversity of ligands it can accommodate may contribute to the large array of roles assigned to PPARγ. The LBP extends from the C-terminal helix H12 to the β-sheet S1/S2 and can be divided into two sub-pockets named hereafter the AF-2 and the β-sheet sub-pockets, respectively57. Based on existing PPARγ complex structures, it has been suggested that full agonists, such as the rosiglitazone (Table 2), occupy both sub-pockets, establishing hydrogen bonds with residues Tyr473 (H12) on one side and Ser342 (S1/S2) on the other side (Figure 4A), whereas partial agonists would bind essentially to the β-sheet sub-pocket58. Interestingly, several structural studies have revealed that the large cavity of PPARγ can accommodate two ligands occupying both AF-2 and β-sheet sub-pockets58,59 (Figure 4B). In addition, we have provided structural and functional evidence that PPARγ can bind simultaneously two or three molecules of the phthalate MEHP and the perfluorinated compound PFOA, respectively (Table 2, unpublished data).

PPARγ bound to pharmaceutical and natural ligands. Close-up views of the ligand-binding pocket of PPARγ in complex with rosiglitazone (A) and the fatty acid 9-HODE (B), showing the two sub-pockets that can be differentially occupied. Oxygen, carbon and nitrogen atoms are shown in red, yellow, and blue, respectively. The dashed lines depict hydrogen bonds.
Organotins and halogenated BPA as potential environmental obesogens
The first environmental obesogen for which a mechanism of action has been elucidated is the organotin tributyltin (TBT, Table 2)60,61. Organotins form a diverse group of synthetic tin derivatives ubiquitously found in the environment due to their widespread use since the 1960s in several industrial and agricultural processes62,63. In the 1980s, these compounds were observed to be responsible for a wide variety of deleterious effects. Despite restrictions on their use, organotins persist in the environment and are absorbed by higher organisms where they accumulate64. Human exposure occurs primarily through diet, in particular through sea food65; however, some organotins have been documented in house dust, suggesting that exposure from sources other than food may be widespread66. TBT and tripropyltin (TPT) are dual nanomolar affinity agonists of PPARγ and its heterodimeric partner RXR and were shown to induce adipogenesis in preadipocytes60,61. A deeper investigation of organotin action indicated that although TBT is a high affinity ligand for both receptors, it acts as a full RXR agonist but activates PPARγ weakly compared to the reference pharmaceutical compound rosiglitazone. Further structural and biophysical studies revealed that TBT binds covalently to both heterodimer subunits by forming a covalent bond between the tin atom and the sulfur atom of cysteine residues located in the LBP of both receptors50 (unpublished data). In RXR, Cys432 is located in helix H11, and although TBT interacts with only a subset of binding pocket residues in the H11–H12 region, it seems to be engaged in enough essential contacts in the AF-2 sub-pocket to stabilize this receptor in its active conformation (Figure 5A). By contrast, the cysteine residue of PPARγ (Cys285) resides in H3 and anchors the organotin in a region of the β-sheet sub-pocket.As a result, it does not allow the efficient stabilization of the active receptor conformation, which is in line with the weak PPARγ agonistic activity of the compound (Figure 5B). Thus, the efficient activation of RXR-PPARγ by TBT results from the combined action of the organotin on the two heterodimer subunits. The discovery of this binding mode suggested that other NRs presenting a cysteine residue in their binding pocket should be considered as potential targets of organotins.The functional outcome of this interaction is dictated by the position of the anchoring cysteine in the LBP. Accordingly, it has been reported that dibutyltin acts as a potent antagonist of the GR, which contains two cysteine residues in its LBP67.

Binding modes of organotins and halogenated bisphenol A. Close-up views of the ligand-binding pocket of RXRα (A) and PPARγ (B, C) in complex with the tributyltin (TBT), tripropyltin (TPT) and tetrabromobisphenol A (TBBPA), respectively. Oxygen, carbon, sulfur, and tin atoms are shown in red, magenta, yellow, and gray, respectively. The dashed lines depict hydrogen bonds. Water molecules are displayed as red spheres.
Halogenated derivatives of BPA, which feature bromine or chlorine substituents on the phenolic rings, are used as flame retardants. Tetrabromobisphenol-A (TBBPA) is used to produce fireproof epoxy resins utilized in the manufacture of computer motherboards and other electronics. Its production is currently estimated to be approximately 200 000 tons/year, and its presence has been reported in the environment68, in wildlife69, and in human samples70,71. The closely related tetrachlorobisphenol-A (TCBPA) is also used as a flame retardant, but in much lower quantities (<10 000 tons/year). Two recent studies have investigated the capacity of the halogenated BPAs and of their biotransformation products to act as PPARγ ligands51,72. Both TBBPA and TCBPA were shown to act as partial agonists of the receptor and promote differentiation of 3T3L1 pre-adipocytes at concentrations within the micromolar range (100-fold less potent than rosiglitazone). These chemicals also activate the corresponding zebrafish and Xenopus receptors51, indicating that halogenated BPA can disrupt the activity of PPARγ from different species. In addition, it was shown that the sulfation pathway, usually considered as a detoxification process, leads to the formation of sulfate conjugates, which possess a significant residual PPARγ binding activity51. The crystal structures of PPARγ in complex with TBBPA and TCBPA are indistinguishable and reveal the canonical tertiary fold of agonist-bound PPAR LBD. The ligands essentially occupy the β-sheet sub-pocket, with one of the phenol rings nestled between H3 and the β-sheet, and only a small part of the AF-2 sub-pocket with no direct interaction with H12 (Figure 5C). However, the second phenol ring of halogenated BPA is indirectly linked to H12 through a water-mediated hydrogen bond with Tyr473. This mode of binding reflects the partial agonistic profiles of the compounds. The four halogen atoms participate in the interaction by establishing van der Waals contacts with the other residues of the LBP. Cell-based assays showed that the activation of PPARγ depends on the halogenation degree of BPA derivatives. The bulkier halogenated BPA analogues, the greater their capability to activate PPARγ. This observation correlates with a higher occupancy of the LBP, resulting in a better stabilization of the active form of the LBD. The crystal structure of the TBBPA metabolite in complex with PPARγ shows that the TBBPA-sulfate is positioned identically to its parent molecule, with the sulfate group filling a portion of the volume occupied by the solvent in the TBBPA structure. The presence of a conserved water molecule mediates the indirect interaction between TBBPA-sulfate and PPARγ helix H12. Overall, the crystallographic data validates the notion that halogenated BPA sulfates are partial agonists of PPARγ.
Other environmental PPARγ ligands
Recent data have revealed that some natural compounds derived from plants can bind to PPARγ and modulate its activity73,74,75. These include RES, luteolin, a flavonoid present in celery, green pepper, perilla leaf and chamomile tea, and magnolol, a lignane, which composes the stem bark of Magnolia officinalis. These compounds exhibit anti-cancer, anti-inflammatory, anti-angiogenic, and/or cardioprotective activities, as well as anti-oxidative effects76,77,78. They bind to PPARγ with micromolar affinities and display weak agonistic activity; magnolol was reported to induce adipogenesis and glucose uptake in 3T3-L1 cells79. It can additionally bind RXRα with a micromolar affinity and was described as a dual agonist of PPARγ and RXRα. Because magnolol agonistic activity can be inhibited by RXR or PPARγ antagonists, magnolol seems to be required on both receptors to positively activate the RXR-PPARγ heterodimer74. As expected from their activity profiles, the crystal structures show that the compounds bind primarily to the β-sheet sub-pocket with no interaction with H12. Moreover, because of their small size and almost planar conformations, these compounds may be found as either two copies or associated with a fatty acid molecule being trapped during purification of the recombinant protein (Figure 6A, 6B).

Differential binding modes of natural and pharmaceutical compounds. Close-up views of the ligand-binding pocket of PPARγ in complex with magnolol (A), luteolin (B), and RU-486 (C). Oxygen, carbon, and nitrogen atoms are displayed in red, magenta, and blue, respectively. The dashed lines depict hydrogen bonds. Water molecules are displayed as red spheres.
The synthetic steroid compound mifepristone (RU-486) is known for its antagonistic properties on the steroid receptors GR and PR and is primarily used for early termination of pregnancy; additionally, it displays an anti-diabetic effect and appears to be a promising drug for the treatment of certain cancers80. RU-486 and its metabolites can be found in the aquatic environment and should be considered as contaminants81. Surprisingly, RU-486 was recently reported to bind to and activate PPARγ82. The structure of the PPARγ LBD in complex with RU-486 shows that PPARγ adopts an active conformation with the steroid occupying the LBP, similar to rosiglitazone (Figure 6C). The steroid core of RU-486 aligns with rosiglitazone and establishes similar hydrophobic interactions and hydrogen bonds with the residues of the LBP, except the critical hydrogen bond with Y473 (H12), which is lost. Furthermore, instead of pointing toward helix H12 as observed in the GR83 and PR84 structures, which account for the antagonistic character of mifepristone, the dimethylaniline side chain of this compound projects towards helix H5 in PPARγ. The difference in the binding modes of RU-486 in steroid receptors and PPARγ explains the divergence in its activities with the two types of receptors.
Conclusion
EDCs are chemicals of great concern because these compounds, which are ubiquitously present in our daily environment, can alter endocrine functions and cause infertility, malformations, metabolic troubles, or increased incidence of cancers. The deregulation of NR-mediated transcription accounts for the deleterious effects of several EDCs, and the weak structural relationships between EDCs and natural hormones render their interaction with these cellular targets poorly understood and barely predictable. Because of these reasons, it is necessary to characterize the harmful interactions between NRs and environmental compounds, at both structural and functional levels, and develop robust in vivo, in vitro, and in silico screening methods.
Using three major biological targets of environmental contaminants (ERα/β and PPARγ) as illustrative examples, the present review highlights the diversity of the mechanisms by which distantly related chemicals bind to and activate NRs with graded efficacy and potency. In addition to providing a better understanding of the differential activities, binding affinities, and specificities of environmental NR ligands, such structural knowledge at a near-atomic level provides rational guidelines to design safer chemicals characterized by fewer NR-mediated side effects. This will increase the effectiveness of 3D-structure based computational tools aimed at predicting the NR-disrupting action of environmental pollutants. Moreover, studies aimed at identifying and characterizing EDCs may aid in finding new ligands for NRs, including orphan receptors, and may reveal unforeseen binding modes, thereby providing guidelines for the rational design of novel NR modulators as new therapeutic agents.
References
Germain P, Staels B, Dacquet C, Spedding M, Laudet V . Overview of nomenclature of nuclear receptors. Pharmacol Rev 2006; 58: 685–704.
Gronemeyer H, Gustafsson JA, Laudet V . Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 2004; 3: 950–64.
Rastinejad F, Huang P, Chandra V, Khorasanizadeh S . Understanding nuclear receptor form and function using structural biology. J Mol Endocrinol 2013; 51: T1–T21.
Li Y, Lambert MH, Xu HE . Activation of nuclear receptors: a perspective from structural genomics. Structure 2003; 11: 741–6.
Bourguet W, Germain P, Gronemeyer H . Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci 2000; 21: 381–8.
Hughes TS, Chalmers MJ, Novick S, Kuruvilla DS, Chang MR, Kamenecka TM, et al. Ligand and receptor dynamics contribute to the mechanism of graded PPARgamma agonism. Structure 2011; 20: 139–50.
Nettles KW, Greene GL . Ligand control of coregulator recruitment to nuclear receptors. Annu Rev Physiol 2005; 67: 309–33.
Nahoum V, Perez E, Germain P, Rodriguez-Barrios F, Manzo F, Kammerer S, et al. Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc Natl Acad Sci U S A 2007; 104: 17323–8.
Bourguet W, de Lera AR, Gronemeyer H . Inverse agonists and antagonists of retinoid receptors. Methods Enzymol 2010; 485: 161–95.
Germain P, Gaudon C, Pogenberg V, Sanglier S, Van Dorsselaer A, Royer CA, et al. Differential action on coregulator interaction defines inverse retinoid agonists and neutral antagonists. Chem Biol 2009; 16: 479–89.
le Maire A, Teyssier C, Erb C, Grimaldi M, Alvarez S, de Lera AR, et al. A unique secondary-structure switch controls constitutive gene repression by retinoic acid receptor. Nat Struct Mol Biol 2010; 17: 801–7.
Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev 2009; 30: 293–342.
Schug TT, Janesick A, Blumberg B, Heindel JJ . Endocrine disrupting chemicals and disease susceptibility. J Steroid Biochem Mol Biol 2011; 127: 204–15.
De Coster S, van Larebeke N . Endocrine-disrupting chemicals: associated disorders and mechanisms of action. J Environ Public Health 2012; 2012: 713696.
le Maire A, Bourguet W, Balaguer P . A structural view of nuclear hormone receptor: endocrine disruptor interactions. Cell Mol Life Sci 2010; 67: 1219–37.
Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, et al. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol Rev 2006; 58: 773–81.
Jensen EV, Jordan VC . The estrogen receptor: a model for molecular medicine. Clin Cancer Res 2003; 9: 1980–9.
Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ . Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006; 126: 789–99.
Couse JF, Korach KS . Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev 1999; 20: 358–417.
Levin ER, Pietras RJ . Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat 2008; 108: 351–61.
Weihua Z, Saji S, Makinen S, Cheng G, Jensen EV, Warner M, et al. Estrogen receptor (ER) beta, a modulator of ERalpha in the uterus. Proc Natl Acad Sci U S A 2000; 97: 5936–41.
Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, et al. Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol Endocrinol 2003; 17: 203–8.
Ellem SJ, Risbridger GP . The dual, opposing roles of estrogen in the prostate. Ann N Y Acad Sci 2009; 1155: 174–86.
Delfosse V, Grimaldi M, Cavailles V, Balaguer P, Bourguet W . Structural and functional profiling of estrogen receptors environmental ligands. Environ Health Perspect 2014; 122: 1306–13.
Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997; 138: 863–70.
Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, et al. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. Embo J 1999; 18: 4608–18.
Nwachukwu JC, Srinivasan S, Bruno NE, Parent AA, Hughes TS, Pollock JA, et al. Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. Elife (Cambridge) 2014; 3: e02057.
Vandenberg LN, Maffini MV, Sonnenschein C, Rubin BS, Soto AM . Bisphenol-A and the great divide: a review of controversies in the field of endocrine disruption. Endocr Rev 2009; 30: 75–95.
Delfosse V, Grimaldi M, Pons JL, Boulahtouf A, le Maire A, Cavailles V, et al. Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes. Proc Natl Acad Sci U S A 2012; 109: 14930–5.
Ikeda K, Arao Y, Otsuka H, Nomoto S, Horiguchi H, Kato S, et al. Terpenoids found in the umbelliferae family act as agonists/antagonists for ER(alpha) and ERbeta: differential transcription activity between ferutinine-liganded ER(alpha) and ERbeta. Biochem Biophys Res Commun 2002; 291: 354–60.
Nettles KW, Sun J, Radek JT, Sheng S, Rodriguez AL, Katzenellenbogen JA, et al. Allosteric control of ligand selectivity between estrogen receptors alpha and beta: implications for other nuclear receptors. Mol Cell 2004; 13: 317–27.
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998; 139: 4252–63.
Lemaire G, Mnif W, Mauvais P, Balaguer P, Rahmani R . Activation of alpha- and beta-estrogen receptors by persistent pesticides in reporter cell lines. Life Sci 2006; 79: 1160–9.
Multigner L, Ndong JR, Giusti A, Romana M, Delacroix-Maillard H, Cordier S, et al. Chlordecone exposure and risk of prostate cancer. J Clin Oncol 2010; 28: 3457–62.
Boucher O, Simard MN, Muckle G, Rouget F, Kadhel P, Bataille H, et al. Exposure to an organochlorine pesticide (chlordecone) and development of 18-month-old infants. Neurotoxicology 2013; 35: 162–8.
Dallaire R, Muckle G, Rouget F, Kadhel P, Bataille H, Guldner L, et al. Cognitive, visual, and motor development of 7-month-old Guadeloupean infants exposed to chlordecone. Environ Res 2012; 118: 79–85.
Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, et al. International union of pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 2006; 58: 726–41.
Varga T, Czimmerer Z, Nagy L . PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta 2011; 1812: 1007–22.
Desvergne B, Wahli W . Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 1999; 20: 649–88.
Berger J, Moller DE . The mechanisms of action of PPARs. Annu Rev Med 2002; 53: 409–35.
Tontonoz P, Spiegelman BM . Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 2008; 77: 289–312.
Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med 2013; 19: 557–66.
Grygiel-Gorniak B . Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications — a review. Nutr J 2014; 13: 17.
Li Y, Zhang J, Schopfer FJ, Martynowski D, Garcia-Barrio MT, Kovach A, et al. Molecular recognition of nitrated fatty acids by PPAR gamma. Nat Struct Mol Biol 2008; 15: 865–7.
Lamers C, Schubert-Zsilavecz M, Merk D . Therapeutic modulators of peroxisome proliferator-activated receptors (PPAR): a patent review (2008-present). Expert Opin Ther Pat 2012; 22: 803–41.
Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010; 466: 451–6.
Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature 2011; 477: 477–81.
Hughes TS, Giri PK, de Vera IM, Marciano DP, Kuruvilla DS, Shin Y, et al. An alternate binding site for PPARgamma ligands. Nat Commun 2013; 5: 3571.
Swedenborg E, Ruegg J, Makela S, Pongratz I . Endocrine disruptive chemicals: mechanisms of action and involvement in metabolic disorders. J Mol Endocrinol 2009; 43: 1–10.
le Maire A, Grimaldi M, Roecklin D, Dagnino S, Vivat-Hannah V, Balaguer P, et al. Activation of RXR-PPAR heterodimers by organotin environmental endocrine disruptors. EMBO Rep 2009; 10: 367–73.
Riu A, Grimaldi M, le Maire A, Bey G, Phillips K, Boulahtouf A, et al. Peroxisome proliferator-activated receptor gamma is a target for halogenated analogs of bisphenol A. Environ Health Perspect 2011; 119: 1227–32.
Janesick A, Blumberg B . Minireview: PPARgamma as the target of obesogens. J Steroid Biochem Mol Biol 2011; 127: 4–8.
Grun F, Blumberg B . Endocrine disrupters as obesogens. Mol Cell Endocrinol 2009; 304: 19–29.
Janesick A, Blumberg B . Obesogens, stem cells and the developmental programming of obesity. Int J Androl 2012; 35: 437–48.
Grun F, Blumberg B . Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 2006; 147 (6 Suppl): S50–5.
Issemann I, Green S . Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990; 347: 645–50.
Zoete V, Grosdidier A, Michielin O . Peroxisome proliferator-activated receptor structures: ligand specificity, molecular switch and interactions with regulators. Biochim Biophys Acta 2007; 1771: 915–25.
Waku T, Shiraki T, Oyama T, Maebara K, Nakamori R, Morikawa K . The nuclear receptor PPARgamma individually responds to serotonin- and fatty acid-metabolites. Embo J 2010; 29: 3395–407.
Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, Balint BL, et al. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat Struct Mol Biol 2008; 15: 924–31.
Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, et al. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol 2006; 20: 2141–55.
Kanayama T, Kobayashi N, Mamiya S, Nakanishi T, Nishikawa J . Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol Pharmacol 2005; 67: 766–74.
Fent K . Ecotoxicology of organotin compounds. Crit Rev Toxicol 1996; 26: 1–117.
Appel KE . Organotin compounds: toxicokinetic aspects. Drug Metab Rev 2004; 36 (3-4): 763–86.
Antizar-Ladislao B . Environmental levels, toxicity and human exposure to tributyltin (TBT)-contaminated marine environment. a review. Environ Int 2008; 34: 292–308.
Fent K . Ecotoxicological problems associated with contaminated sites. Toxicol Lett 2003; 140-141: 353–65.
Kannan K, Takahashi S, Fujiwara N, Mizukawa H, Tanabe S . Organotin compounds, including butyltins and octyltins, in house dust from Albany, New York, USA. Arch Environ Contam Toxicol 2010; 58: 901–7.
Gumy C, Chandsawangbhuwana C, Dzyakanchuk AA, Kratschmar DV, Baker ME, Odermatt A . Dibutyltin disrupts glucocorticoid receptor function and impairs glucocorticoid-induced suppression of cytokine production. PLoS ONE 2008; 3: e3545.
de Wit CA, Herzke D, Vorkamp K . Brominated flame retardants in the Arctic environment — trends and new candidates. Sci Total Environ 2009; 408: 2885–918.
Darnerud PO . Toxic effects of brominated flame retardants in man and in wildlife. Environ Int 2003; 29: 841–53.
Cariou R, Antignac JP, Zalko D, Berrebi A, Cravedi JP, Maume D, et al. Exposure assessment of French women and their newborns to tetrabromobisphenol–A: occurrence measurements in maternal adipose tissue, serum, breast milk and cord serum. Chemosphere 2008; 73: 1036–41.
Shi H, Qian L, Guo S, Zhang X, Liu J, Cao Q . Teratogenic effects of tetrabromobisphenol A on Xenopus tropicalis embryos. Comp Biochem Physiol C Toxicol Pharmacol 2009; 152: 62–8.
Riu A, le Maire A, Grimaldi M, Audebert M, Hillenweck A, Bourguet W, et al. Characterization of novel ligands of ERalpha, Erbeta, and PPARgamma: the case of halogenated bisphenol A and their conjugated metabolites. Toxicol Sci 2011; 122: 372–82.
Calleri E, Pochetti G, Dossou KS, Laghezza A, Montanari R, Capelli D, et al. Resveratrol and its metabolites bind to PPARs. Chembiochem 2014; 15: 1154–60.
Zhang H, Xu X, Chen L, Chen J, Hu L, Jiang H, et al. Molecular determinants of magnolol targeting both RXRalpha and PPARgamma. PLoS One 2011; 6: e28253.
Puhl AC, Bernardes A, Silveira RL, Yuan J, Campos JL, Saidemberg DM, et al. Mode of peroxisome proliferator-activated receptor gamma activation by luteolin. Mol Pharmacol 2012; 81: 788–99.
Fremont L . Biological effects of resveratrol. Life Sci 2000; 66: 663–73.
Xagorari A, Papapetropoulos A, Mauromatis A, Economou M, Fotsis T, Roussos C . Luteolin inhibits an endotoxin-stimulated phosphorylation cascade and proinflammatory cytokine production in macrophages. J Pharmacol Exp Ther 2001; 296: 181–7.
Havsteen BH . The biochemistry and medical significance of the flavonoids. Pharmacol Ther 2002; 96: 67–202.
Choi SS, Cha BY, Lee YS, Yonezawa T, Teruya T, Nagai K, et al. Magnolol enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Life Sci 2009; 84: 908–14.
Chen J, Wang J, Shao J, Gao Y, Xu J, Yu S, et al. The unique pharmacological characteristics of mifepristone (RU486): from terminating pregnancy to preventing cancer metastasis. Med Res Rev 2014 Sep; 34: 979–1000.
Scott PD, Bartkow M, Blockwell SJ, Coleman HM, Khan SJ, Lim R, et al. An assessment of endocrine activity in Australian rivers using chemical and in vitro analyses. Environ Sci Pollut Res Int 2014; 21: 12951–67.
Lin S, Han Y, Shi Y, Rong H, Zheng S, Jin S, et al. Revealing a steroid receptor ligand as a unique PPARgamma agonist. Cell Res 2012; 22: 746–56.
Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M, et al. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem 2003; 278: 22748–54.
Raaijmakers HC, Versteegh JE, Uitdehaag JC . The X-ray structure of RU486 bound to the progesterone receptor in a destabilized agonistic conformation. J Biol Chem 2009; 284: 19572–9.
Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997; 389: 753–8.
Manas ES, Xu ZB, Unwalla RJ, Somers WS . Understanding the selectivity of genistein for human estrogen receptor-beta using X-ray crystallography and computational methods. Structure 2004; 12: 2197–207.
Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, et al. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol 2008; 4: 241–7.
Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998; 95: 927–37.
Mocklinghoff S, Rose R, Carraz M, Visser A, Ottmann C, Brunsveld L . Synthesis and crystal structure of a phosphorylated estrogen receptor ligand binding domain. Chembiochem 2010; 11: 2251–4.
Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998; 395: 137–43.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Delfosse, V., Maire, A., Balaguer, P. et al. A structural perspective on nuclear receptors as targets of environmental compounds. Acta Pharmacol Sin 36, 88–101 (2015). https://doi.org/10.1038/aps.2014.133
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2014.133
Keywords
This article is cited by
-
Natural compounds targeting nuclear receptors for effective cancer therapy
Cancer and Metastasis Reviews (2023)
-
Risk of thyroid cancer and benign nodules associated with exposure to parabens among Chinese adults in Wuhan, China
Environmental Science and Pollution Research (2022)
-
Insights into the activation mechanism of human estrogen-related receptor γ by environmental endocrine disruptors
Cellular and Molecular Life Sciences (2019)
-
Confirmation of high-throughput screening data and novel mechanistic insights into VDR-xenobiotic interactions by orthogonal assays
Scientific Reports (2018)
-
Identification of a novel selective PPARγ ligand with a unique binding mode and improved therapeutic profile in vitro
Scientific Reports (2017)
