
Abstract
Cellular senescence is defined as the physiological program of terminal growth arrest, which can be triggered by various endogenous or exogenous stress signals. Cellular senescence can be induced in response to oncogenic activation and acts as a barrier to tumorigenesis. Moreover, tumor cells can undergo senescence when exposed to chemotherapeutic agents. In addition to suppressing tumorigenesis, senescent cells remain metabolically active and may contribute to tumor formation and to therapy resistance. In the current review, we discuss the molecular regulation of cellular senescence, the potential implications of senescence in human cancers, and the possibility of exploiting cellular senescence for the treatment of cancers.
Similar content being viewed by others
Introduction
Cellular senescence was originally defined as the proliferative arrest that occurs in normal cells after a number of cell divisions, even upon treatment with mitogens. This process of “replicative senescence” in normal cells results primarily from the shortening of, and other structural changes to, telomeres at the ends of chromosomes1. Cellular senescence was first described 50 years ago by Hayflick and Moorehead2 and was called the “Hayflick limit”. Senescent cells typically appear flattened and enlarged and show increased cytoplasmic granularity. In addition to the characteristic changes in morphology, senescent cells display several other differences from quiescent cells. These differences include senescence-associated β-galactosidase activity (SA-β-gal), senescence-associated heterochromatin foci (SAHF), and high expression levels of p16 and p213, 4, 5. These senescence markers can be used to identify senescent cells both in vitro and in vivo. In addition to replicative senescence, cellular senescence can be induced by other types of stress, such as oncogene activation (Figure 1)6. The oncogenic protein Ras and its effectors, such as activated mutant RAF, MEK, and BRAF, have all been shown to cause senescence7, 8, 9. This form of cellular senescence is similar to the Hayflick limit; however, in contrast to Hayflick's observations, Ras-induced senescence is much more rapid and is independent of telomere shortening. Courtois-Cox and colleagues have referred to this form of cellular senescence as oncogene-induced senescence (OIS)9. Many laboratories have reported that OIS may act as a barrier to neoplastic transformation, and bypass of OIS is a prerequisite for tumorigenesis. Recent studies also suggest a link between OIS and induced-pluripotency in in vitro settings, supporting the idea that cellular senescence counteracts the induced-conversion of primary cells into pluripotent stem cells10, 11, 12. Thus, OIS may suppress tumor formation not only by inducing a persistent cell-cycle arrest, but also by limiting the generation of cancer stem cells. Therapy-induced senescence (TIS) is another form senescence that refers to the phenomenon of a subset of tumor cells being forced into a senescent state by therapeutic agents (Figure 1). The ability of therapeutic stimuli to induce tumor cell senescence has been noted for different treatments, including radiation and chemotherapeutic drugs (such as doxorubicin and cisplatin)13, 14. These observations indicate that cancer cells harbor signaling pathways/mechanisms that can be utilized to induce senescence. Recently, the ability of cancer cells to overcoming TIS has been proposed as one mechanism behind cancer recurrence and drug resistance. However, how TIS contributes to cancer recurrence and drug resistance remains an unanswered question15, 16. Although only certain therapeutics can induce senescence, and senescence does not occur in all the treated cells, TIS has clinical implications and significance in regard to the efficacy and effectiveness of treatment regimens. In the current review, we discuss the molecular regulation of the aforementioned forms of cellular senescence, the possible clinical implications of senescence in human cancer, and the potential for exploiting cellular senescence for the treatment of cancer.

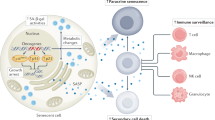
Induction of cellular senescence by various stimuli. Cells can activate intrinsic pathways to undergo replicative senescence (RS), oncogene-induced senescence (OIS) or therapy-induced senescence (TIS).
Replicative senescence and targeting of telomeres/telomerase in cancer
Telomeres and replicative senescence
The phenomenon of replicative senescence (RS) was first observed in primary human cells that had a finite life span in cell culture. These primary human cells grew in culture but stopped dividing after a number of divisions17. RS mainly occurs in response to dysfunctional telomeres. Telomeres become slightly shorter after each cellular division and are eventually too short to allow the cell to divide, resulting in cellular senescence and apoptosis. Telomerase can prevent telomere erosion and the subsequent cellular senescence in highly proliferating cells. The dependence of replicative senescence on telomere shortening is evident, based on the fact that senescence can be bypassed by telomerase reverse transcriptase (hTERT), a catalytic subunit that elongates telomeres. In the presence of hTERT, RS is significantly reduced18. The majority of cancerous cells (>90%) express telomerase to maintain telomere length19, 20; however, cancer cells can also elongate their telomere through another mechanism termed alternative lengthening of telomeres (ALT)20. This alternative mechanism was discovered in some cancer cell lines in which telomere lengths were maintained in the absence of telomerase activity16. Thus, human cancer cells maintain their telomeres and consequently the ability to proliferate indefinitely making telomeres and telomerase ideal targets for therapeutic intervention in combating cancer.
Telomerase, telomeres, and cancer therapy
Various therapeutic strategies targeting telomeres and telomerase have been developed, including gene therapy, immunotherapy, telomerase inhibitors and telomere-disrupting agents (Figure 2). Antisense gene therapies in which the hTERT mRNA or telomerase mRNA are targeted with RNAi or hammerhead ribozymes have been shown to selectively impact telomerase-positive cells. Such agents include antisense oligonucleotides, peptide nucleic acids (PNAs), and chemically modified PNAs such as GRN163L. GRN163L has been reported to inhibit telomerase activity in cancer cells, thereby promoting telomere shortening and subsequently cell cycle arrest and apoptosis21. To date, most studies have focused on searching for and testing natural agents or synthesizing chemical compounds that inhibit telomerase activity in cancer cells, resulting in loss of the telomere maintenance mechanism and induction of senescence and apoptosis. Several compounds, such as BIBR1532 {2-[(E)-3-naphtalen-2-yl-but-2-enoylamino]-benzoic acid}22 and TNQX (2,3,7-trichloro-5-nitroquinoxaline)23, 24, have been reported to possess these properties. Protein kinase C (PKC) and Akt have been shown to phosphorylate hTERT and to enhance telomerase activity25. Therefore, it is conceivable that inhibitors of these protein kinases could have inhibitory effects on telomerase activity. Recently, a different approach to targeting telomerase has been reported. Ugel and colleagues developed an adoptive cellular therapy by expanding high-avidity T cells reactive against telomerase epitopes. When these cells were injected back into transgenic mice, adenocarcinoma progression was hampered. A similar strategy was also effective against human cancer. In a mouse model, T cells reactive against human telomerase successfully abolished tumor growth of several types of human cancer cells26. Clinical trials are currently going on to evaluate these approaches, and it will be interesting to see whether or not these approaches will work in cancer patients. Currently, all of these approaches target telomerase and telomeres; no specific inhibitor has yet been designed to inhibit the ALT pathway. ALT-mediated telomere maintenance may underlie the observed resistance of some tumor cells to telomere-based therapies. This presents a fascinating area for future investigation, as combining ALT and telomere-based therapies may greatly improve outcomes for these therapies.

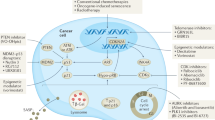
Pathways regulating RS and targeting RS for cancer therapy. RS mainly occurs in response to dysfunctional telomeres, which is the result of telomere erosion or telomere shortening. In cancer cells, telomerase or alternative lengthening of telomeres (ALT) can prevent telomere erosion or telomere shortening and subsequent cellular senescence. Various strategies to targeting telomeres and telomerase have been developed, including hTERT immunotherapy, telomerase inhibitors, telomere-disrupting agents and some protein kinase inhibitors.
Oncogene-induced senescence and its role in cancer treatment and development
Cellular senescence can also be induced by oncogenes, and this type of senescence is termed oncogene-induced senescence9. Unlike RS, the induction of OIS is much more rapid and cannot be bypassed by the expression of hTERT, indicating that OIS is independent of telomere attrition27. OIS is triggered by abnormal signaling within a cell, which is mediated by the protein products of oncogenes and is often accompanied by activation of tumor-suppressor networks. Critical signaling pathways shared by both RS and OIS are the p53 and p16/pRB (retinoblastoma tumor suppressor) pathways. Some oncogenes such as RAS, STAT5, and cyclin E not only induce senescence but also trigger a DNA-damage response, which is associated with DNA hyper-replication and appears to be involved in OIS28. It is now becoming increasingly clear that OIS is a crucial anticancer mechanism that prevents the growth of cells that are at risk for neoplastic transformation. Conversely, oncogenic stimuli that induce senescence also have the potential to initiate tumor promotion (Figure 3).

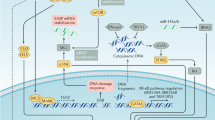
Pathways regulating OIS and neoplastic transformation. Primary human cells that acquire active oncogenes undergo OIS. When tumor suppressor genes are lost or oncogenes are over-expressed, cells bypass OIS and are eventually transformed. The most common pathways involved in OIS are the p53 and Rb pathways. OIS may also be induced by other mechanisms, such as secretion of senescence-inducing proteins, autophagy and miRNAs. Oncogenic stimuli that induce a senescence response may also have the potential to initiate tumor promotion.
Effector pathways and therapy
Signaling pathways known to regulate OIS include the p16/pRb and p19/p53/p21 pathways. Reduction of p53 function by dominant negative mutants, specific p53 antisense mRNA, oligonucleotides, or viral oncoproteins is sufficient to extend the lifespan of several cell-types in culture29. The protein MDM2 has p53 ubiquitin ligase activity and is part of the autoregulatory feedback loop that regulates p53 activity. Overexpression of MDM2 targets p53 for degradation and results in loss of p53 function30. Furthermore, the human homologue of MDM2 is frequently overexpressed in melanoma31, suggesting that alternative mechanisms for p53 inactivation occur in human cancers. Studies have demonstrated that restoring p53 activity by suppressing MDM2 overexpression (eg, by nutlin treatment) can achieve clinical benefits in patients with melanoma or other types of cancer. Activation of p53 induces the expression of p21, a cyclin-dependent kinase (CDK) inhibitor that has a direct inhibitory effect on cell cycle progression. In human cells, depletion of p21 is sufficient to bypass senescence32. However, in mouse embryonic fibroblasts, knockout of p21 does not overcome senescence33, 34, suggesting that at least one additional downstream effector is needed for p53-induced senescence. Other proteins such as 14-3-3 and GADD45 are also believed to be involved in modulating cellular senescence35, 36.
The retinoblastoma tumor suppressor, Rb, also plays a regulatory role in senescence. Overexpression of Rb or overexpression of various regulators of the pRb pathway such as CDK inhibitors leads to a senescent phenotype37. CDKs are enzymes that stimulate cell cycle progression by phosphorylating the Rb protein (pRb). One such CDK inhibitor is p16, which tightly binds to Rb to control its major effector, E2F, thereby preventing DNA replication and entry into the S phase of the cell cycle38. Cross-talk between effectors involved in OIS has also been revealed. For instance, myc, a gene that is involved in oncogenic signaling pathways, is able to induce cells to bypass senescence. Myc can bypass CDK4/6 inhibition by activating CDK-2-cyclin A/E and by up-regulating the CDK-activated phosphatase, cdc25A39. Therefore, inhibition of the CDKs involved in these epigenetic modifications could lead to senescence and may have anti-cancer activity. CDKs can be inhibited by using adenoviral vectors carrying CKIs (CDK inhibitors such as p16, p21, p15, and p27)40. Further studies are needed to determine the potential of this approach as an anticancer strategy.
Secreted proteins in OIS
A major consequence of oncogene activation is the secretion of dozens of proteins, including cytokines and chemokines41. Some of these secreted proteins and their receptors are known to be regulators of OIS. For example, CXCR2 (interleukin-8 receptor type-B) was identified as an important regulator of OIS, which can lead to growth inhibition42. In addition, Wajapeyee and colleagues43 performed a genome-wide RNA interference screening and found that the secreted protein, IGFBP7, plays a central role in BRAFV600E-mediated senescence and apoptosis. Expression of BRAFV600E in primary cells leads to synthesis and secretion of IGFBP7, which acts through autocrine/paracrine pathways to inhibit BRAF-MEK-ERK signaling and to induce senescence and apoptosis. Nickoloff and colleagues reported that the tumor-suppressive secreted protein, maspin is up-regulated in senescent tumor cells. Maspin expression is also increased in aging skin keratinocytes in vivo. The upregulation of maspin in senescent keratinocytes was found to result in a secreted anti-angiogenic activity detectable using an in vitro assay. However, the pool of proteins secreted in response to oncogenes is complex, and some components are actually tumor-promoting. For example, it has been reported that the secreted protein Trefoil factor 1 (TFF1) allows human prostate epithelial cells to escape OIS caused by activation of the Ras oncogene or by reduced expression of the tumor suppressor PTEN44. Yang and colleagues45 revealed that pre-neoplastic epithelial ovarian cells express GRO1 (a cytokine recognized by CXCR2), which is secreted in response to signaling by the RAS oncogene and has the potential to induce senescence in ovarian fibroblasts in vitro. Furthermore, these senescent ovarian fibroblasts promoted tumor growth when co-injected with pre-neoplastic epithelial cells into mice. Senescent cells also secrete high levels of IL-6, IL-8, VEGF, and MMPs, which can stimulate the migration and invasion of pre-malignant epithelial cells46, 47, 48, 49. These studies indicate that senescent tumor cells have the ability to secrete factors that inhibit the growth of their non-senescent neighbors. However, senescent tumor cells may also secrete factors that promote proliferation of tumor cells that reside in their microenvironment. Recent studies have presented the following paradox: while a senescent response is generally associated with tumor suppression, some senescent cells can produce a second-wave of secreted proteins that facilitate and drive cancer development. A clear picture of factors contributing to this paradox will require a deeper understanding of how the genes encoding secreted proteins are up-regulated as a consequence of oncogene activation. It may be possible to selectively inhibit some of the senescence-induced secretory proteins (eg, VEGF or MMPs) to abrogate tumor beneficial effects, which could be a strategy to harness the power of intrinsic tumor-suppressive mechanisms for improving cancer therapies.
Autophagy in OIS
Autophagy is a crucial cellular process involved in maintaining cellular homeostasis by helping cells cope with metabolic stress and limiting oxidative damage. Defective autophagy has been linked to various human diseases including cancer, neurodegeneration, and aging. Interestingly, both autophagic flux and expression of regulators of autophagy are increased in OIS cells50, and de-regulation of H-Ras activity can lead to caspase-independent cell death with features of autophagy. Modulation of key autophagic components such as Ulk3, Atg5, or Atg7 has been shown to alter cellular senescence. This effect may occur through feedback control of the PI3K-Akt-mTOR pathway, which acts to limit oncogenic signaling and enables cell cycle exit51. These studies suggest that the pathways involved in autophagy and senescence could be functionally linked. Autophagy has apparently conflicting roles in the development and maintenance of cancer. On the one hand, activators of autophagy likely inhibit transformation and prevent cancer by limiting cellular damage. On the other hand, autophagy plays an important role in the growth and survival of pancreatic cancer cells52. Although autophagy is induced in response to oncogene activation, this cellular process can also be induced in response to a variety of other stresses such as starvation or hypoxia. It remains to be determined whether the autophagy process is the same in response to different activators. Induction of Ulk3 has been shown to be sufficient to stimulate autophagy and OIS53. The induction of Ulk3 by OIS is associated with induction of a set of autophagy-related genes that is distinct from that induced by starvation or hypoxia, indicating that the autophagic response induced in OIS might be a distinct form of autophagy that facilitates the process of OIS. Defining what aspects of autophagy are required for maintaining the senescent phenotype and for suppressing tumor formation are important questions for future investigation.
MicroRNAs in OIS
MicroRNAs are a class of evolutionarily conserved non-coding RNAs that regulate the expression of protein-encoding genes54, 55. In recent years, numerous miRNAs have been identified and implicated in human cancer56, 57, 58. Not surprisingly, miRNAs also play important roles in the regulation of senescence and oncogenesis. For example, miR-17-92 was reported to inhibit oncogenic RAS-induced senescence and to promote oncogenesis59, including evasion of apoptosis58. Overexpression of miR-380 cooperates with active H-RAS oncoproteins to transform primary cells, block oncogene-induced senescence, and form tumors60. Using genome-wide siRNA and miRNA screening, Borgdorff and colleagues showed that 28 miRNAs (miR-17-5p, miR-130b, etc) were involved in preventing RASG12V-induced senescence in human mammary epithelial cells (HMECs)61. Similarly, two miRNAs (miR-372 and miR-373) have been shown to rescue Ras-induced senescence in human fibroblasts and have been found to be overexpressed in testicular germ cell tumors62. Importantly, these miRNAs have also been linked to cancer development. For example, miR-17-5p is overexpressed in pancreatic cancer63, breast cancer64, hepatocellular cancer65, and thyroid cancer66, while miR-130b is overexpressed in gastric cancer67. These observations demonstrate the roles of miRNAs in oncogenic RAS-induced senescence and apoptosis, and suggest that regulation of senescence by miRNAs might be part of a universal mechanism of miRNA-mediated growth control. Therefore, induction of senescence by miRNA-based therapy might be a promising approach for the prevention and treatment of cancer. Recent therapeutic applications of miRNAs include the following two major strategies: 1) For oncogenic miRNAs that are overexpressed in tumors and promote cell proliferation, the therapeutic approach would be to suppress miRNA expression. This could be accomplished by using anti-miRNA oligonucleotides, miRNA masking, and small molecule inhibitors of miRNAs. 2) For tumor-suppressor miRNAs (TS-miRNAs), which are often lost in tumors, the therapeutic strategy would be to restore expression of TS-miRNAs by exogenous transfection of designated miRNA mimics. Some miRNA-based therapeutic molecule delivery methods have been developed and applied in clinical trials, including viral and non-viral vector systems.
Therapy-induced senescence (TIS), a double-edged sword?
As mentioned above, the progression to malignancy involves bypassing or inhibiting crucial mediators of cellular senescence. Nevertheless, this does not mean that malignant cells have completely lost their capacity to undergo senescence. Given the intrinsic tumor-suppressive potential of senescence, investigation of the effectiveness of senescence-inducing interventions for the treatment of cancer should be interesting and potentially useful. Therapy-induced senescence (TIS) is another form of cellular senescence that has been described recently. Although TIS is caused by genotoxic agents such as irradiation and chemotherapeutic drugs, it shares many similarities to OIS (eg, common markers) (Figure 4).

TIS, a double-edged sword? In response to therapeutic intervention, cancer cells can rapidly undergo apoptosis or enter TIS. These senescent cells can be cleared by the immune system, which is beneficial for the host. In contrast, some cancer cells in a senescence-like state can remain 'dormant' or possess a secretory phenotype. They therefore possess a dangerous potential for tumor relapse, which is considered a detrimental outcome for the host.
Induction of senescence in vitro and in vivo by chemotherapy
A variety of anticancer agents can induce senescence-like morphological changes and SA-β-gal expression in tumor cells. These drugs include the topoisomerase I inhibitor camptothecin68, the topoisomerase II inhibitor doxorubicin69, the DNA damaging agents cisplatin and doxorubicin70, γ-irradiation71, and the anti-metabolite cytarabine72. These observations suggest that different drugs may activate senescence through different signaling pathways. DNA damage response pathways have been shown to play a critical role in OIS, RS, and TIS. For instance, the DNA methyltransferase inhibitor, 5-azacytidine, has been shown to cause senescence in DU145 and LNCaP prostate cancer cells within 7 d after treatment73. Sirtinol, an inhibitor of histone DNA acetylase, induced senescence in MCF7 breast and H1299 lung cancer cells after 24 h of exposure to this agent74. Recently, the involvement of the c-Jun N-terminal kinase p38 (MAPK 14) and the extracellular signal-regulated kinase (MAPK 1) pathways in TIS have also been reported75, 76.
TIS has also been observed in vivo. Two recent reports77, 78 have addressed the pro-senescence efficacy of restoring the tumor suppressive function of p53. Most of the sarcomas, lymphomas and liver carcinomas that developed in the absence of p53 regressed after p53 restoration. These reports support the idea that drugs that restore p53 function in tumors may provide an effective means of using senescence to restrict tumor growth. Several p53-restoring agents are currently being developed, such as MI-219 and Tenovin-6. In addition, Alimonti and colleagues reported that the PTEN inhibitor, VO-OHpic, induced a novel type of cellular senescence in mouse models of prostate cancer79. Wouters, BG and colleagues reported that tumor cells with altered levels of the CDK inhibitor and growth arrest gene CIP1 produced similar results in clonogenic assays following γ-irradiation but appeared to be more radio-sensitive in the absence of p21 in vivo. This suggests that genetic protection from γ-irradiation-induced growth arrest may result in a higher rate of remission if apoptosis is still available80. Using newly sectioned specimens from frozen breast tumors of patients treated with the chemotherapeutic agents doxorubicin, 5-fluorouracil and cyclophophamide, Poele and colleagues81 observed SA-β-gal staining in tumor cells but not in normal tissue. They further found that SA-β-gal staining in breast cancer was associated with low p53 staining and high p16 staining, suggesting that chemotherapy-induced senescence is a specific response of tumor cells. In a similar study, 2 out of 3 tumor biopsies from patients treated with carboplatin/paclitaxel were positive for markers of cellular senescence, whereas no significant SA-β-gal activity was found in 3 tumor samples obtained from untreated patients82. These results suggest that clinical benefits might be achieved by neoadjuvant chemotherapy for various cancer entities as the tumor lesions present at the time of surgery may not only be largely senescent thus reducing tumor size, but such therapy could also limit the growth potential of the remaining masses.
Escape of tumor cells from TIS
It is conceivable that some cancer cells in a senescence-like state might remain 'dormant' or possess tumor stem cell characteristics, therefore representing a dangerous potential for tumor relapse. If drug-inducible senescence is indeed of clinical importance, then mechanisms that allow tumor cells to overcome this terminal arrest might contribute to anticancer drug resistance and tumor relapse. In fact, some in vitro experiments have already demonstrated that senescent cells can re-enter the mitotic cycle upon acquisition of additional senescence-compromising mutations83, 84, 85. In vivo observations also suggest that escaping from drug-induced senescence is responsible, at least in part, for treatment failure. For example, Braig and colleagues86 observed in mice harboring drug-induced senescent tumors that loss of genes critical for the induction and maintenance of cellular senescence was facilitated by a heterozygotic state for the genes in question; however, mice heterozygous at those alleles also appeared to be particularly prone to overcoming drug-induced arrest.
Clinical applications of cellular senescence-based therapy: is it a parable?
TIS has several features that may be beneficial for the management and treatment of cancer. First, senescence stimulates a persistent terminal growth arrest and senescent cells may persist in a stable state even in vivo. For example, senescent melanocytes have been identified in benign nevi that remain dormant for years6. It has also been reported that a subset of senescent prostate cancer cells persisted for at least 6 weeks after establishment of xenografts with doxorubicin-induced senescent prostate cancer cells87. Second, induction of senescence in tumor cells can stimulate an immune response, which increases susceptibility to cytotoxicity through lymphokine-activated killer cells88. It has been observed in a mouse liver carcinoma model that re-expression of a functional p53 induced senescence, resulting in tumor regression89, 90. This response may also be instrumental in the improved survival observed in a mouse lymphoma model in which senescence was induced by chemotherapy and BCL290. Third, the observation that senescence can be induced by lower concentrations of chemotherapeutic agents suggests another potential benefit of TIS for cancer treatment. Schwarze and colleagues73 tested a range of concentrations of doxorubicin, 5-azacytidine, and docetaxel for the ability to induce senescence and found that, at lower concentrations, the senescent phenotype was predominant in most prostate cancer cells, whereas higher concentrations led to DNA damage and were associated with apoptosis. These studies demonstrated that induction of senescence in tumors may be achieved with lower concentrations of therapeutic agents, which may help limit drug-related toxic effects.
Cellular senescence is generally considered to be irreversible in most cases, thus making it an attractive therapeutic target for diseases like cancer. Nevertheless, we must pay close attention to the potential problems that might arise from senescence-inducing therapies. The reversibility of senescence, particularly in fibroblasts, cannot yet be ruled out, as the factors involved in maintenance of senescence may stop being expressed83, 84. This possibility remains to be further clarified experimentally. In addition, the occurrence of cellular senescence may result in resistance to cell death programs such as apoptosis, autophagic cell death or necrosis. For example, senescent fibroblasts are resistant to the apoptotic effects of serum starvation91 and hydrogen peroxide92. Pro-apoptotic signaling via ceramide and TNF-α can be blocked in the senescent fibroblasts. However, whether TIS causes resistance of cancer cells to apoptosis has not been clearly demonstrated. Additionally, as mentioned above, senescent cells have a robust secretory phenotype, which results in the production of pro-inflammatory cytokines and chemokines. These may also stimulate malignant phenotypes in nearby tumor cells93. For example, Chang and colleagues94 found that chemotherapeutic agent-induced senescence of HCT116 cells not only led to up-regulation of growth inhibitors but also increased the levels of secreted factors with tumor-promoting activity. Such secreted factors included extracellular matrix components, Cyr61 and prosaposin (anti-apoptotic factors), transforming growth factor α (TGFα), and several proteases that could potentially contribute to metastatic growth. Coppe and colleagues47 also found that senescent fibroblasts secrete pro-inflammatory immune cytokines, including IL-6 and IL-8, which have the potential to promote bystander cell proliferation and may account for the development of some cancers. In this regard, it is important to gain a deeper understanding of the mechanisms responsible for the clearance of senescent tumor cell.
Conclusions and perspectives
Senescence-inducing agents, when combined with other classic therapeutic approaches, may prove to be more effective in the treatment of cancer. Thus, gaining a better understanding of the importance and significance of cellular senescence and exploring how to modulate this cellular process to treat cancer have become areas of extensive research. The advantages of enhancing senescence include terminal growth arrest, low toxicity-related side effects, and immune stimulation. However, under certain conditions, acutely-induced senescence has been shown to be a reversible program. Moreover, cellular senescence may contribute to resistance to the cytotoxic effects of cancer therapies. Senescent cells have the potential to promote the proliferation of bystander cells and may thus account for the development of certain types of cancer. To make the best use of the intrinsic tumor-suppressive potential of cancer cells, further studies are needed to determine whether senescence is indeed relevant to cancer development and therapeutic responses. Future studies should focus on searching for and developing robust senescence-inducing agents, identifying more reliable senescence markers, discovering new regulators of cellular senescence, and designing new therapeutic strategies that force transformed cancer cells to enter the senescence program and promote clearance of these senescent cells.
References
Campisi J, d'Adda di Fagagna F . Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729–40.
Hayflick L, Moorhead PS . The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25: 585–621.
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92: 9363–7.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW . Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88: 593–602.
Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, et al. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006; 126: 503–14.
Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005; 436: 720–4.
Chandeck C, Mooi WJ . Oncogene-induced cellular senescence. Adv Anat Pathol 2010; 17: 42–8.
Braig M, Schmitt CA . Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res 2006; 66: 2881–4.
Courtois-Cox S, Jones SL, Cichowski K . Many roads lead to oncogene-induced senescence. Oncogene 2008; 27: 2801–9.
Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev 2009; 23: 2134–9.
Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009; 460: 1140–4.
Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009; 460: 1145–8.
Wang X, Wong SC, Pan J, Tsao SW, Fung KH, Kwong DL, et al. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res 1998; 58: 5019–22.
Di Leonardo A, Linke SP, Clarkin K, Wahl GM . DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 1994; 8: 2540–51.
Cesinaro AM, Migaldi M, Ferrari G, Castagnetti G, Dotti A, De Gaetani C, et al. Expression of p53 and Bcl-2 in clinically localized prostate cancer before and after neo-adjuvant hormonal therapy. Oncol Res 2000; 12: 43–9.
Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR . Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J 1995; 14: 4240–8.
Hayflick L . The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965; 37: 614–36.
Vaziri H, Benchimol S . Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 1998; 8: 279–82.
Shay JW, Bacchetti S . A survey of telomerase activity in human cancer. Eur J Cancer 1997; 33: 787–91.
Muntoni A, Reddel RR . The first molecular details of ALT in human tumor cells. Hum Mol Genet 2005; 14: R191–6.
Harley CB . Telomerase and cancer therapeutics. Nat Rev Cancer 2008; 8: 167–79.
Pascolo E, Wenz C, Lingner J, Hauel N, Priepke H, Kauffmann I, et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J Biol Chem 2002; 277: 15566–72.
Colangelo D, Ghiglia A, Ghezzi A, Ravera M, Rosenberg E, Spada F, et al. Water-soluble benzoheterocycle triosmium clusters as potential inhibitors of telomerase enzyme. J Inorg Biochem 2005; 99: 505–12.
Strahl C, Blackburn EH . The effects of nucleoside analogs on telomerase and telomeres in Tetrahymena. Nucleic Acids Res 1994; 22: 893–900.
Kim MO, Moon DO, Kang SH, Heo MS, Choi YH, Jung JH, et al. Pectenotoxin-2 represses telomerase activity in human leukemia cells through suppression of hTERT gene expression and Akt-dependent hTERT phosphorylation. FEBS Lett 2008; 582: 3263–9.
Ugel S, Scarselli E, Iezzi M, Mennuni C, Pannellini T, Calvaruso F, et al. Autoimmune B-cell lymphopenia after successful adoptive therapy with telomerase-specific T lymphocytes. Blood 2010; 115: 1374–84.
Wei S, Sedivy JM . Expression of catalytically active telomerase does not prevent premature senescence caused by overexpression of oncogenic Ha-Ras in normal human fibroblasts. Cancer Res 1999; 59: 1539–43.
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444: 638–42.
Wynford-Thomas D . p53: guardian of cellular senescence. J Pathol 1996; 180: 118–21.
Blaydes JP, Wynford-Thomas D . The proliferation of normal human fibroblasts is dependent upon negative regulation of p53 function by mdm2. Oncogene 1998; 16: 3317–22.
Polsky D, Bastian BC, Hazan C, Melzer K, Pack J, Houghton A, et al. HDM2 protein overexpression, but not gene amplification, is related to tumorigenesis of cutaneous melanoma. Cancer Res 2001; 61: 7642–6.
Rowland BD, Bernards R, Peeper DS . The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol 2005; 7: 1074–82.
Takeuchi S, Takahashi A, Motoi N, Yoshimoto S, Tajima T, Yamakoshi K, et al. Intrinsic cooperation between p16INK4a and p21Waf1/Cip1 in the onset of cellular senescence and tumor suppression in vivo. Cancer Res 2010; 70: 9381–90.
Zhao B, Benson EK, Qiao R, Wang X, Kim S, Manfredi JJ, et al. Cellular senescence and organismal ageing in the absence of p21CIP1/WAF1 in ku80−/− mice. EMBO Rep 2009; 10: 71–8.
Kuroda Y, Aishima S, Taketomi A, Nishihara Y, Iguchi T, Taguchi K, et al. 14-3-3sigma negatively regulates the cell cycle, and its down-regulation is associated with poor outcome in intrahepatic cholangiocarcinoma. Hum Pathol 2007; 38: 1014–22.
Jackson JG, Pereira-Smith OM. p53 is preferentially recruited to the promoters of growth arrest genes p21 and GADD45 during replicative senescence of normal human fibroblasts. Cancer Res 2006; 66: 8356–60.
Haddad MM, Xu W, Schwahn DJ, Liao F, Medrano EE . Activation of a cAMP pathway and induction of melanogenesis correlate with association of p16(INK4) and p27(KIP1) to CDKs, loss of E2F-binding activity, and premature senescence of human melanocytes. Exp Cell Res 1999; 253: 561–72.
Liu Y, El-Naggar S, Clem B, Chesney J, Dean DC . The Rb/E2F pathway and Ras activation regulate RecQ helicase gene expression. Biochem J 2008; 412: 299–306.
Amati B, Alevizopoulos K, Vlach J . Myc and the cell cycle. Front Biosci 1998; 3: d250–68.
Carnero A . Targeting the cell cycle for cancer therapy. Br J Cancer 2002; 87: 129–33.
Campisi J . Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2002; 120: 513–22.
Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008; 133: 1006–18.
Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR . Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008; 132: 363–74.
Radiloff DR, Wakeman TP, Feng J, Schilling S, Seto E, Wang XF . Trefoil factor 1 acts to suppress senescence induced by oncogene activation during the cellular transformation process. Proc Natl Acad Sci U S A 2011; 108: 6591–6.
Yang G, Rosen DG, Zhang Z, Bast RC Jr, Mills GB, Colacino JA, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A 2006; 103: 16472–7.
Coppe JP, Kauser K, Campisi J, Beausejour CM . Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem 2006; 281: 29568–74.
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6: 2853–68.
Ksiazek K, Jorres A, Witowski J . Senescence induces a proangiogenic switch in human peritoneal mesothelial cells. Rejuvenation Res 2008; 11: 681–3.
Parrinello S, Coppe JP, Krtolica A, Campisi J . Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci 2005; 118: 485–96.
Patschan S, Chen J, Polotskaia A, Mendelev N, Cheng J, Patschan D, et al. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am J Physiol Heart Circ Physiol 2008; 294: H1119–29.
Elgendy M, Sheridan C, Brumatti G, Martin SJ . Oncogenic ras-induced expression of noxa and beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell 2011; 42: 23–35.
Yang S, Wang X, Contino G, Liesa M, Sahin E, ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25: 717–29.
Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev 2009; 23: 798–803.
Bartel DP . MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–33.
Shi XB, Tepper CG, deVere White RW . Cancerous miRNAs and their regulation. Cell Cycle 2008; 7: 1529–38.
Dillhoff M, Wojcik SE, Bloomston M . MicroRNAs in solid tumors. J Surg Res 2009; 154: 349–54.
Croce CM . Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 2009; 10: 704–14.
Hong L, Lai M, Chen M, Xie C, Liao R, Kang YJ, et al. The miR-17-92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res 2010; 70: 8547–57.
Swarbrick A, Woods SL, Shaw A, Balakrishnan A, Phua Y, Nguyen A, et al. miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat Med 2010; 16: 1134–40.
Borgdorff V, Lleonart ME, Bishop CL, Fessart D, Bergin AH, Overhoff MG, et al. Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21Waf1/Cip1. Oncogene 2010; 29: 2262–71.
Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006; 124: 1169–81.
Yu J, Ohuchida K, Mizumoto K, Fujita H, Nakata K, Tanaka M . MicroRNA miR-17-5p is overexpressed in pancreatic cancer, associated with a poor prognosis, and involved in cancer cell proliferation and invasion. Cancer Biol Ther 2010; 10: 748–57.
O'Day E, Lal A . MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res 2010; 12: 201.
Yang F, Yin Y, Wang F, Wang Y, Zhang L, Tang Y, et al. miR-17-5p Promotes migration of human hepatocellular carcinoma cells through the p38 mitogen-activated protein kinase-heat shock protein 27 pathway. Hepatology 2010; 51: 1614–23.
Takakura S, Mitsutake N, Nakashima M, Namba H, Saenko VA, Rogounovitch TI, et al. Oncogenic role of miR-17-92 cluster in anaplastic thyroid cancer cells. Cancer Sci 2008; 99: 1147–54.
Yao Y, Suo AL, Li ZF, Liu LY, Tian T, Ni L, et al. MicroRNA profiling of human gastric cancer. Mol Med Report 2009; 2: 963–70.
Hayward RL, Macpherson JS, Cummings J, Monia BP, Smyth JF, Jodrell DI . Antisense Bcl-xl down-regulation switches the response to topoisomerase I inhibition from senescence to apoptosis in colorectal cancer cells, enhancing global cytotoxicity. Clin Cancer Res 2003; 9: 2856–65.
Elmore LW, Rehder CW, Di X, McChesney PA, Jackson-Cook CK, Gewitz DA, et al. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem 2002; 277: 35509–15.
Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, et al. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res 1999; 59: 3761–7.
Mirzayans R, Scott A, Cameron M, Murray D . Induction of accelerated senescence by gamma radiation in human solid tumor-derived cell lines expressing wild-type TP53. Radiat Res 2005; 163: 53–62.
Rosenbeck L, Kiel PJ . Images in clinical medicine. Palmar-plantar rash with cytarabine therapy. N Engl J Med 2011; 364: e5.
Schwarze SR, Fu VX, Desotelle JA, Kenowski ML, Jarrard DF . The identification of senescence-specific genes during the induction of senescence in prostate cancer cells. Neoplasia 2005; 7: 816–23.
Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 2006; 25: 176–85.
Hong EH, Lee SJ, Kim JS, Lee KH, Um HD, Kim JH, et al. Ionizing radiation induces cellular senescence of articular chondrocytes via negative regulation of SIRT1 by p38 kinase. J Biol Chem 2010; 285: 1283–95.
Chen JY, Hwang CC, Chen WY, Lee JC, Fu TF, Fang K, et al. Additive effects of C(2)-ceramide on paclitaxel-induced premature senescence of human lung cancer cells. Life Sci 2010; 87: 350–7.
Martinelli P, Bonetti P, Sironi C, Pruneri G, Fumagalli C, Raviele PR, et al. The lymphoma-associated NPM-ALK oncogene elicits a p16INKa/pRb-dependent tumour-suppressive pathway. Blood 2011; 117: 6617–26.
Chao SK, Lin J, Brouwer-Visser J, Smith AB 3rd, Horwitz SB, McDaid HM . Resistance to discodermolide, a microtubule-stabilizing agent and senescence inducer, is 4E-BP1-dependent. Proc Natl Acad Sci U S A 2011; 108: 391–6.
Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, Trotman LC, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest 2010; 120: 681–93.
Wouters BG, Giaccia AJ, Denko NC, Brown JM . Loss of p21Waf1/Cip1 sensitizes tumors to radiation by an apoptosis-independent mechanism. Cancer Res 1997; 57: 4703–6.
te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP . DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 2002; 62: 1876–83.
Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY . Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res 2005; 65: 2795–803.
Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22: 4212–22.
Dirac AM, Bernards R . Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem 2003; 278: 11731–4.
Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T . Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 2003; 424: 223–8.
Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005; 436: 660–5.
Ewald J, Desotelle J, Almassi N, Jarrard D . Drug-induced senescence bystander proliferation in prostate cancer cells in vitro and in vivo. Br J Cancer 2008; 98: 1244–9.
Petti C, Molla A, Vegetti C, Ferrone S, Anichini A, Sensi M . Coexpression of NRASQ61R and BRAFV600E in human melanoma cells activates senescence and increases susceptibility to cell-mediated cytotoxicity. Cancer Res 2006; 66: 6503–11.
Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445: 656–60.
Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002; 109: 335–46.
Wang E . Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 1995; 55: 2284–92.
Sasaki M, Kumazaki T, Takano H, Nishiyama M, Mitsui Y . Senescent cells are resistant to death despite low Bcl-2 level. Mech Ageing Dev 2001; 122: 1695–706.
Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev 2010; 29: 709–22.
Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB . Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci U S A 2002; 99: 389–94.
Acknowledgements
This article was supported by the US Public Health Service R01CA135038 and the National Natural Sciences Foundation of China 81072146.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zhang, Y., Yang, Jm. The impact of cellular senescence in cancer therapy: is it true or not?. Acta Pharmacol Sin 32, 1199–1207 (2011). https://doi.org/10.1038/aps.2011.108
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.108
Keywords
This article is cited by
-
P53-dependent downregulation of hTERT protein expression and telomerase activity induces senescence in lung cancer cells as a result of pterostilbene treatment
Cell Death & Disease (2017)
-
Friend or foe: the role of microRNA in chemotherapy resistance
Acta Pharmacologica Sinica (2013)
-
Oxidative stress in apoptosis and cancer: an update
Archives of Toxicology (2012)
