
Abstract
The year of 2005 was a watershed in the history of poly(ADP-ribose) polymerase (PARP) inhibitors due to the important findings of selective killing in BRCA-deficient cancers by PARP inhibition. The findings made PARP inhibition one of the most promising new therapeutic approaches to cancers, especially to those with specific defects. With AZD2281 and BSI-201 entering phase III clinical trials, the final application of PARP inhibitors in clinic would come true soon. This current paper will review the major advances in targeting PARP for cancer therapy and discuss the existing questions, the answers to which may influence the future of PARP inhibitors as cancer therapeutics.
Similar content being viewed by others
Introduction
In 2005, two groups of Bryant HE, et al1 and Farmer H, et al2 reported their discovery of highly selective anticancer activity of poly(ADP-ribose) polymerase (PARP) inhibition in BRCA1- and BRCA2-deficient cancers in the same issue of Nature. Subsequent successful clinical trials with PARP inhibitors for cancer therapy have proved that the discovery made a major breakthrough at least at two aspects: raising a very promising selective anticancer strategy and vigorously boosting the research and development of PARP inhibitors as cancer therapeutics. This current review will be focused on these two aspects, especially on the new advances and existing questions in the field of anticancer PARP inhibitors due to several brilliant comprehensive reviews available3, 4, 5.
Targeting PARPs, a promising selective anticancer strategy
PARPs related to the activity of DNA
The first member of the PARP superfamily, PARP1, was reported in 19636 and up to now, this superfamily has expanded to at least 17 members including PARP1, PARP2, PARP3, PARP4/vPARP, PARP5a/tankyrase 1 (Tank1) and PARP5b/tankyrase 2 (Tank2)4. Although all are characteristic of a common conserved catalytic domain, only five members (PARP1, PARP2, PARP4, Tank1, and Tank2) possess the bona fide poly(ADP-ribosyl)ation activity of transferring and polymerizing ADP-ribose from nicotinamide adenine dinucleotide (NAD+) onto acceptor proteins3, 7, 8. The others reveal only the transferase activity of transferring mono(ADP-ribose) (MAR), with the exception of PARP9 and PARP13, two of which show no identified enzymatic activity7. In fact, poly(ADP-ribosyl)ation and mono(ADP-ribosyl)ation of proteins have been shown to function differently. The former forms long branched polymers of PAR on targeted proteins, which provide scaffolds to further molecular reactions, to regulate the actions of the modified proteins, or to promote their degradation; the latter adds a single MAR to its targeted proteins, the activity of which may thus be changed7, 8. As an example, PARP3 mono(ADP-ribosyl)ates PARP1 and activates the latter even at the condition of no DNA binding that is required for the autoactivation of PARP1 itself8.
The members in the PARP superfamily related to the activity of DNA mainly include PARP1, PARP2, Tank1, and Tank2. Within the cell, PARPs exert differential functions due to their different abundance, targeting proteins and cellular locations. PARP1 accounts for at least 80% of total cellular PARP activity9, and together with its nearest relative PARP2, constitutes the DNA-damage-dependent PARPs9, 10. Mainly located in the nucleus, PARP1 poly(ADP-ribosyl)ates various nuclear proteins including histones H1 and H2B, XRCC1, CENP-A, CENP-B, BUB3, and itself, whereas PARP2 adds PAR to XRCC1, CENP-A, CENP-B, BUB3, and itself11, 12, 13. Most of those proteins are involved in the activities of DNA and centrosomes, especially in DNA-damage repair, specifically the base excision repair (BER). PARP1/2 function as DNA single strand break (SSB) sensors. Once one strand of DNA breaks, PARP1/2 bind to the nicked site, which activates their own enzymatic activity, causing the poly(ADP-ribosyl)ation of PARP1/2 and other nuclear proteins including histones and XRCC1 at the expense of NAD+. Consequently, DNA polymerase β and the ligase III-XRCC1 complex are recruited to the poly(ADPribosyl) ated PARP1/2 to repair the SSB14, 15 .
Though associated with genomic stability, Tank1 and Tank2 act in a different way from PARP1/2. Both tankyrases are found at telomeres where they facilitate telomere elongation by poly(ADP-ribosyl)ating the negative regulator of telomere length, TRF1, and then releasing it from the telomeres16, 17. Tank1 associates with TRF1 and regulates telomere length. In fact, cells deficient in Tank1 suffer a block in resolution of sister telomeres and arrest in early anaphase due to a persistent telomere association18. Tank1 is thus believed to disrupt a “telomere cohesion complex” that holds telomeres together till separation at anaphase18. Consistent with the in vitro observations, both Tank1 and Tank2 are found to be essential though redundant for mouse embryonic development19. In addition, Tank1/2 are also found to regulate the Wnt/β-cantenin pathway (see the following). These findings highlight a novel important function of poly(ADP-ribosyl)ation.
In addition, some members of the PARP superfamily may also indirectly affect the activity of DNA via the mono(ADP-ribosyl)ation of PARP1 as by PARP3 or of histones as by PARP3 and PARP107, 8 . However, the precise biological functions of most of the other PARPs remain largely unclear.
Targeting PARP as a selective anticancer strategy
Targeting PARP 1/2
PARP1/2 are essential to repair SSB. Deficiency in the PARP1/2 activity leads to the accumulation of SSB. When colliding the progressing replication forks, the SSB are converted to DNA double-strand breaks (DSB)20, 21. To repair such DSB, the homologous recombination (HR) pathway and the ATM-Chk1 signaling pathway must be activated22. Deficiency in both the pathways, ie functional mutations in their critical components (for example, BRCA1/2, ATM and Chk1) disables the HR repair, which confers lethality to those DSB.
This forms the basic principle for cancer therapy by targeting PARP1/2. Apart from the inhibition of PARP1/2, in this principle, another two prerequisites are SSB and aberrant HR repair. SSB can be endogenous and exogenous. Each day, about 20 000 DNA lesions per cell result from all endogenous DNA damage such as spontaneous DNA depurination, damage from reactive oxygen species, deamination of bases and alkylation damage23. Among those lesions, a significant fraction is or will be converted to SSB that requires PARP1/2 for repair. Exogenous SSB can be generated with ionizing radiation, alkylating agents (for example, temozolomide, methyl-methanesulfonate (MMS), cyclophosphamide), platinum (cisplatin, carboplatin, oxaliplatin) and topoisomerase I inhibitors (topotecan, irrinotecan)3, 24. Generating the endogenous and exogenous SSB forms the basis for selective and combination anticancer treatments, respectively.
On the other hand, aberrant HR repair mainly results from the defects in the HR pathway and the ATM-Chk1 pathway. Both BRCA1 and BRCA2 are an essential component of a core HR complex14. Their inactivated mutations make the HR repair lose its function and predispose individuals to cancers due to genomic instability. In fact, the inactivation of BRCA1 and BRCA2 is responsible for nearly 10% of all invasive breast cancers20 and for a fraction of ovarian and prostate cancers. Moreover, a life risk of breast cancer is greater than 80% in those carriers of BRCA1 or BRCA2 mutations25. But not only that, the abnormality in BRCA1 is also related to colorectal cancer and in BRCA2, even more extensively, to cancers at buccal cavity and pharynx, stomach, pancreas, melanoma of the skin, and gallbladder and bile ducts26, 27, 28. In contrast, both ATM and Chk1 are important kinases in DNA damage signaling, responsible for damage sensing and modulating cell cycle progression in order to facilitate the HR repair. Consistently, deficiency in ATM and Chk1 impairs the HR repair and is associated with leukemia and various other cancers22, 24.
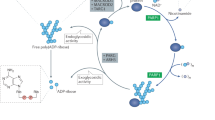
Therefore, inhibition of PARP1/2 in BRCA1- and BRCA2 -deficient cancers results in synthetic lethality due to the disabled repair for daily endogenous DNA damage1, 2, 3, 29 (Figure 1). In this situation, targeting PARP1/2 elicits selective anticancer effects by only killing the cancer cells with two mutated copies of those genes but sparing the normal cells with one good copy of them1, 2, 29. This strategy thus distinguishes malignant cells from normal cells based on their differential BRCA, totally different from the current strategies in clinic by using cytotoxic drugs or molecular-targeted drugs. In fact, cytotoxic drugs non-selectively kill cancer and normal cells due to targeting the essential biological components including DNA, topoisomerases and tubulin to both cell types. Similarly though a little slightly, the present molecular-targeted drugs, targeting biological molecules such as EGFR, VEGFR, and PDGFR that are differentially activated in both cancer and normal cells, also afflict the normal cells because those molecules are essential to the cells, especially in those rapid-turnover tissues. Another different point is that all BRCA1, BRCA2, and ATM function as tumor suppressors while the molecules (ie, EGFR, VEGFR, PDGFR, etc) 30 targeted by the present molecular-targeted drugs generally play a role of oncogenes in cancers. Therefore, targeting PARP1/2 is, in nature, to indirectly target the defects of cancers, and in theory, faces much lower risk of systematic toxicity than using conventional cytotoxic drugs or the present molecular-targeted drugs.

A schematic presentation of the rationale of targeting PARPs for cancer therapy. Inhibition of PARPs impairs the repair for DNA single-strand breaks (SSB), which then accumulate in the cell. When colliding the progressing replication forks, those SSB are converted to DNA double-strand breaks (DSB) that are generally repaired through the homologous recombination (HR) pathway. Defects in the HR pathway such as the functional mutations of BRCA1 or BRCA2 disable the repair for those DSB and confer lethality to them. This is so called as “synthetic lethality”. On the other hand, if the HR pathway functions normally under PARP inhibition, additional introduction of exogenous SSB as induced by irradiation or certain DNA damaging drugs may produce excessive DSB that overwhelm the HR repair. Consequently, those excessive, unrepaired DSB kill the cell.
In BRCA1- and BRCA2-proficient cancers, inhibition of PARP1/2 may potentiate anticancer effects of inducing exogenous SSB, for example, with ionizing radiation, oxaplatin or irinotecan3, 24. In this occasion, too many exogenous SSB are accumulated in the cell subsequent to PARP1/2 inhibition and are converted to DSB, which probably overwhelm the normal HR repair and finally become lethal (Figire 1). Because both PARP1/2 inhibition and exogenous SSB induction are nonselective for cancer and normal cells, this approach renders a relatively high risk of systematic toxicity though likely achieving significant chemopotentiation31. Consistent with this theory, accumulating nephrotoxicity of cisplatin was observed when in combination with the PARP inhibitor AZD228132.
Targeting Tank1/2
Although the functions of Tank1/2 remain to be thoroughly clarified, both have revealed a possibility to act as anticancer targets at least in two different ways: one is via the telomere-telomerase system and the other is via the Wnt/β-catenin pathway. In the former, the telomere-specific DNA binding protein TRF1 is a critical mediator of the effect of targeting Tank1/2. Normally, TRF1 binds to the telomere, which prevents telomerase from its access to the telomere33, 34. Tank1/2 poly(ADP-ribosyl)ate TRF1 and release it from the telomere and force it to degradation via the ubiquitin-proteasome pathway, consequently facilitating the telomerase-mediated elongation of the telomere16, 35, 36. Inhibition of Tank1/2 increases the binding of TRF1 to the telomere and thus accelerates the telomere shortening37. Therefore, Tank1/2 inhibition may potentiate the telomere shortening due to telomerase inhibition and circumvent the cellular resistance to telomerase inhibitors.
In the latter, the β-catenin suppressor Axin plays a role in relaying the effect of targeting Tank1/2 to the Wnt/β-catenin pathway, very similar to the role of TRF1 in mediating the effect of targeting Tank1/2. Axin is the scaffold of the Axin complex consisting of GSK3α/β and CKIα and APC, which phosphorylating β-catenin and making it degraded through the ubiquitin-proteasome pathway38. Recently, Axin has been shown to be poly(ADP-ribosyl)ated by Tank1/2 followed by being ubiquitinated and degraded also via the proteasome pathway39, 40. As a result, Tank1/2 may, through promoting the degradation of the suppressor (Axin) of β-catenin, enhance the activity of the Wnt/β-catenin pathway. Inhibition of Tank1/2 prevents Axin from the poly(ADP-ribosyl)ation-mediated degradation, increases its own levels, and thus reduces β-catenin by promoting its destruction, finally inhibiting the Wnt/β-catenin pathway39, 40.
Because both the telomere-telomerase system and the Wnt/β-catenin pathway are abnormally overactivated in various cancers, targeting Tank1/2 could achieve, to some extent, selective anticancer effects. In this case, there are at least 3 impressive features in targeting Tank1/2. Firstly, both pathways are controlled negatively by a corresponding specific factor (ie, TRF1 or Axin) that is further regulated negatively by Tank1/2. Therefore, targeting Tank1/2 is, in essence, to remove its own inhibition on a negative regulator and thus to enhance the latter's suppression on the relevant overactivated pathway. Such a mode of targeting “dual-inhibition” is interesting and unique though its anticancer effectiveness remains to be further evaluated. Secondly, targeting Tank1/2 prevents TRF1 or Axin from being degraded via the poly(ADP-ribosyl)ation-dependent ubiquitin-proteasome pathway. This Tank1/2-executed poly(ADP-ribosyl)ation triggers the degradation (of TRF1 or Axin) but not activates them, prettily different from targeting PARP1/2. Finally, targeting Tank1/2 concurrently inhibits two important pathways, which may elicit enhanced anticancer effects in the cancers with overactivation of those pathways.
PARP inhibitors as cancer therapeutics
Since 2005, the rapid development of PARP inhibitors for cancer therapy has brought at least 7 PARP inhibitors into clinical anticancer investigations, and 2 of them have entered phase III clinical trials for cancer therapy. The encouraging results from those clinical studies further reveal a promising future of targeting PARP as an anticancer strategy.
PARP inhibitors: classification and their molecular modes of action
The current PARP inhibitors can be classified based on the type of the targeted PARPs and on their chemical structure and mode of action. According to the targeted PARPs, PARP inhibitors can be divided into the inhibitor of PARP1, PARP2, Tank1, Tank2, and so on. Among them, only PARP1/2 inhibitors are in clinical trials for cancer therapy, and only two Tank1/2 inhibitors40 are shown a preliminary potential as cancer therapeutic agents.
PARP inhibitors fall into competitive and noncompetitive inhibitors according to whether they contain the nicotinamide pharmacophore and compete the catalytic site of the enzyme with nicotinamide5. The reported inhibitors, with very few exceptions, belong to the competitive inhibitors. In fact, only two, BSI-201 [4-iodo-3-nitro-benzamide, (INO2BA)] and BSI-401 [6-iodo-5-amino-1,2- benzopyrone], containing no nicotinamide pharmacophore, represent the noncompetitive ones. Both eject the zinc from the first zinc-finger of the PARP1 protein and thus inactivate the enzyme41, 42. In addition, BSI-201 inactivates PARP1 by completely digesting the PARP1 protein itself via activating a PARP1 protease41, 43. BSI-201 is one of the most specific and effective C-nitroso-derivative PARP1 inhibitors so far, showing a significant tumoricidal effect against different cancer cell lines, especially when in combination of the glutathione-synthesis inhibitor buthionine sulfoximine41, 43. BSI-401 also exhibited prominent antitumor activity when used alone or in combination with oxaliplatin44.
The competitive PARP inhibitors, most of which are PARP1/2 inhibitors, play a leading role in current drug development programs. Such inhibitors are designed to mimic nicotinamide and compete for the catalytic domain of PARPs5, 45, 46, 47, 48. For PARP1 inhibitors, as yet, more than 30 scaffolds have been reported, including isoquinolinones, dihydroisoquinolinones, quinazolinediones, quinazolinones, benzimidazole and benzoxazole carboxamides (detailed in the comprehensive review by Ferraris DV)5. Based on them, thousands of derivatives are produced. The X-ray cocrystals of the chicken PARP1 and its inhibitors revealed the binding modes of those lactams45. The amide group of each inhibitor forms three hydrogen bonds in the nicotinamide binding pocket of the enzyme protein, one from Ser904 and two from Gly863 in the nicotinamide binding site45. Any substitution close to the amide usually impairs the potency of its PARP1 inhibition due to disrupting those critical hydrogen bonds. In contrast, two aryl residues, Tyr896 and Tyr907 in the same pocket, form π-π stack with the flat arylamide of the inhibitor, which potentiates its PARP1 inhibition; for this reason, inhibitors with saturated amides generally possess reduced activities.
However, such an approach to mimic nicotinamide generally yields inhibitors with relatively low specificity for different isoforms, specifically PARP1 and PARP2, for the binding environment between these PARPs is virtually identical45, 46, 49. In this case, off-target effects are almost unavoidable. Nevertheless, to decrease the off-target effects and also to investigate the specific effect of each PARP isoenzyme, some isoenzyme-specific PARP inhibitors have been developed and reported, for example, PARP2 inhibitors (UPF-1035, UPF-1069)50 . Undoubtedly, these isoform specific inhibitors can become sharp tools in exploring the differential functions of PARP isoenzymes in the cell. But their potential implications in cancer therapy remain to be further evaluated because of the redundant functions between those isoforms, just as PARP1 and PARP2 in the SSB repair and Tank1 and Tank2 in the telomere-telomerase system and in the Wnt/β-catenin pathway.
Clinical development of PARP inhibitors as cancer therapeutics
Before 2005, PARP inhibitors were mainly tested in combination with other anticancer agents such as irradiation, temozolomide and oxaliplatin for their radio- or chemo-potentiation. But after 2005, the concept of sythetical lethality conceived from the studies of Bryant HE, et al1 and Farmer H, et al2 forcefully stimulated the trials of PARP inhibitors alone for their selective anticancer activity in BRCA1- and BRCA2-deficient cancers.
Clinical trials with single-agent PARP inhibitors in treating BRCA1- and BRCA2-deficient cancers
Using genetic interference and PARPA1/2 inhibitors, Bryant HE, et al1 and Farmer H, et al2 independently found that PARP1/2 inhibition resulted in 100–1000 times greater killing in BRCA-deficient cancers than in BRCA-proficient cells. This finding directly conceived the concept of sythetical lethality and targeting the defects of cancers, which moved the focus of the PARP-inhibitor development from other diseases (ie, cardiac ischemia, inflammation) onto cancers, especially those with the HR-repair deficiency. All current traceable clinical trials with 7 PARP inhibitors [BSI-201(BiPar/Sanofi, phase III*), AZD2281 (KuDOS/AstraZeneca, phase III*), ABT-888 (Abbot, phase II*), AG -014699 (Pfizer, phase II*), INO-1001 (Inotek, phase II*), CEP-9722 (Cephalon, phase I*) and MK-4827 (Merck, phase II*)] are designed to treat cancers (* represents the most advanced stage)5. Several of them, ie, BSI-201, AZD2281, ABT-888, and AG-014699, are being evaluated for their single-agent therapy in BRCA-deficient breast and/or ovarian cancers, showing encouraging therapeutic effects with least systematic toxicity.
As a novel, potent, and orally bioavailble PARP inhibitor, AZD2281 as monotherapy was reported to achieve prominent therapeutic results in BRCA-associated cancers in a phase I study51. Among 60 evaluated patients, 23 carried the BRCA1- or BRCA2- mutated cancers but only 19 (15, ovarian; 3, breast; 1, prostate) had evaluable clinical responses. Among them, treatment with AZD2281 at 400 mg or 600 mg produced a clinical benefit of 63% (12/19) characteristic of stabilization or tumor regression, and 47% (9/19) had a partial or complete response with the response in one patient persistent for over 76 weeks51. Notably, AZD2281, at the conditions of producing the objective therapeutic responses, only led to very limited, reversible toxicities, which were basically grade 1–2 adverse events51. Following this phase I study, its phase II clinical trials have also been completed (but the results have not been reported yet) and now AZD2281 is undergoing its phase III clinical evaluations5.
Clinical trials of PARP inhibitors in combination with DNA damaging agents
Though the monotherapy as with AZD2281 in clinical trials shows an inspiring prospect in populations with cancers of specific defects, but such specific populations are too small, limiting the clinical development of PARP inhibitors. On the other hand, the efficacy of the monotherapy based on the limited daily endogenous DNA damage may be actually potentiated by introduction of additional exogenous DNA damage as generated with irradiation and some DNA damaging anticancer drugs32, 52, 53. To expand the potential clinical indications and also to enhance the therapeutic efficacy, clinical development of some PARP inhibitors are going back to the mode of combination with DNA damaging agents that had been explored prior to the monotherapy. Actually, most of ongoing clinical trials with PARP inhibitors are designed in a combination therapy manner.
A phase II clinical trial with the PARP1 inhibitor BSI-201 in combination with gemcitabine/carboplatin was completed last year in 116 patients with triple-negative metastatic breast cancer (TNBC)54. This clinical evaluation shows a definite clinical benefit in the patients of roughly 62% receiving both BSI-201 and chemotherapy, in striking contrast to the response of only 21% in the patients receiving chemotherapy alone. Moreover, the introduction of BSI-201 greatly prolonged the median lifespan up to 9.2 months as compared with 5.7 months in the control groups53, 55, 56. Based on these encouraging data, a phase III clinical trial of BSI-201 has been started in July 2009 (NCT00938652). Characteristic of negative estrogen receptor, progesterone receptor and HER2, of the BRCA mutations and of high metastatic potential, TNBC defies almost all current hormone-based and HER2-targeted therapies57. The situation strongly urges new therapeutic approaches to TNBC that accounts for up to 10%−20% of all breast cancers58. And similar situation also exists in ovarian cancers. Therefore, the clinical results of BSI-201 indeed blazed a trail in those refractory cancers with high lethality.
As a matter of fact, PARP inhibitors are also clinically tested for an expansion of cancer types and anticancer agents (http://clinicaltrials.gov). In addition to breast, ovarian and prostate cancers, many other types of advanced malignancies are also under clinical investigation including squamous-cell lung cancer, non-small-cell lung cancer, malignant glioma, uterus tumor, peritoneal cancer, gastric cancer, colon tumor, pancreatic cancer, metastatic malignant melanoma, and lymphoma/leukemia. The anticancer agents in combination also range from SSB-inducing agents including DNA alkylating agent (temozolomide, MMS, dacarbazine), topoisomerase I inhibitors (topotecan, irinotecan), DNA cross-linking agents (cisplatin, oxaliplatin and carboplatin) and ionizing radiation, to other DNA damaging agents such as topoisomerase II inhibitors (doxorubicin), nucleoside analogues (gemcitabine), and antifolates (pemetrexed), and to non-DNA-damaging agents just as microtubulin inhibitors (paclitaxel, vinorelbine) and even bevacizumab, a monoclonal antibody against vascular endothelial growth factor. Those clinical trials may, if successfully, remarkably expand the future application of PARP inhibitors. So the results are urgently waited for by all the related parties including researchers, physicians and pharmaceutical companies.
Existing questions in the field of PARP inhibitors and their possible answers
Though a very explicit promise are emerging from the clinical trials, there still exist numerous questions remaining to be solved in order to accelerate the development of PARP inhibitors as cancer therapeutics, especially related to the functions of PARPs, designing and application of their inhibitors.
On the functions of PARPs
At least 17 members of the PARP superfamily have been reported. However, only PARP1 has been relatively thoroughly investigated for its distribution at subcellular, cellular and tissue levels, its modes of action, its functions and its roles in diseases. In particular, its essential role in the SSB repair has been clarified well and for this reason, the field of targeting PARP for cancer therapy has been opened. PARP2 is the second most understood member in this superfamily that is redundant with PARP1 in their function of DNA repair but distinct from the latter in some other aspects. For example, PARP2 is deeply involved in the regulation of the immunological system21, 59, 60. Additionally, Tank1 and Tank2 are found to have redundant functions in regulating the telomere-telomerase system and the Wnt/β-catenin pathways36, 39. Many other members of this superfamily have been least reported for their biological functions, needless to say their roles in diseases.
This scenario brings forth the following two critical questions that are necessary to be answered in developing better PARP inhibitors as cancer therapeutics. (1) Do we need specific PARP inhibitors, say, specific PARP1, PARP2, or Tank1, Tank2 inhibitors? The embryonic lethality of double (but not single) knockout mice of PARP1/PARP2 or Tank1/Tank2 indicates their redundant functions and suggests a necessity of concurrently targeting PARP1 and PARP2 or Tank1 and Tank2 for effective cancer killing. In contrast, inhibition of PARP1, but not PARP2, specific killed BRCA2-deficient tumors, suggesting specific PARP1 inhibitor may be fewer side effects than co-inhibitors of PARP1/21. Indeed, the non-specific PARP1/2 inhibitors AZD2281, AG014699, and ABT-888 were reported to cause the dose-limiting toxicity of myelosuppression whereas the specific PARP1 inhibitor BSI-201 revealed its lack of normal tissue toxicity though similar anticancer effects were achieved31. This difference seems to suggest that specifically targeting PARP1 is a better choice though it is difficult to exclude the impact of the different administration regimens in those studies31. So it becomes important to developing inhibitors of specific PARPs to comparatively test their therapeutic effects and systematic toxicity in clinic. (2) Can any other members in this superfamily except for PARP1 and PARP2 be targeted for cancer therapy? According to very preliminary experimental evidence39, 40, Tank1 and Tank2 could be such ones but need to be further validated. For the other members, more studies are required.
On the designing of new PARP inhibitors
Most of the PARP inhibitors under clinical trials reveal very potent in vitro activity of PARP1/2 inhibition with IC50 ranging from 1 nmol/L to 6 nmol/L, and in humans, result in more than 90% inhibition of the PARP1/2 enzymatic activity31, 51. Therefore, efforts just focused on how to enhance the potency or efficacy of enzymatic inhibition may only achieve limited success in improving the benefit of patients though such a strategy has continuously been ameliorated31. So what can we do? To answer this question, we first need to examine what the problems of the current PARP inhibitors are. Practically, the current PARP inhibitors are mainly faced with two problems: one is the therapeutic effectiveness due to its strict dependency on the deficient HR and endogenous DNA damage when used alone, and the other is the possible increase in the systematic toxicity when in combination with DNA damaging agents. So, these two must be borne in mind in designing new PARP inhibitors. To tackle the first problem, one may design agents concurrently targeting PARP1/2 and the critical components in the HR repair or in the DSB signaling pathway, for example, ATM or Chk1, and such inhibitors can be called “dual functional inhibitors” of the SSB and DSB repairs. This strategy may be feasible because PARP inhibitors and ATM or Chk1 inhibitors are actually synergistic29, 61, 62. The latter appears to be more difficult to deal with, and thus we may turn to more careful selection of the DNA damaging agents or even the non-DNA damaging agents in combination according to the specific types of cancers.
Additionally, the mode of action of the PARP1 inhibitor BSI-201 seems very special and notable. According to the earliest report on this agent41, BSI-201 only showed relatively weak in vitro enzymatic inhibition of PARP1 with an IC50 of about 100 μmol/L. However, it shows excellent anticancer effects with very low systematic toxicity either preclinically or clinically5. Distinct from other competitive PARP inhibitors, BSI-201 does not compete the catalytic domain of the PARP1 enzyme with nicotinamide41. Otherwise, it is possible that BSI-201 may inhibit the binding of PARP1 to the DNA single-strand broken ends that are known as the activator of the enzymatic activity of PARP1. If this is true (though needing additional experimental evidence), targeting the allosteric site of PARPs or the binding of PARPs to the DNA and thus preventing their activation by SSB may be another selectable strategy in designing new inhibitors. In addition to directionally designing new compounds, for this purpose, it could be valuable to introduce natural products (because of their diversity of chemical structures) and their derivatives into the corresponding drug-discovery programs, thus obtaining some unexpected active compounds for further optimization. This may require modifying the current mode of PARP inhibitor screening and evaluation mainly based on determining enzymatic activity.
On the application of PARP inhibitors for cancer therapy
Preclinical and clinical anticancer investigations show that PARP inhibitors, though promising, have questions yet to be answered as follows. (1) To what degree of PARP inhibition can the ideal anticancer therapeutic effects be achieved in the cell and especially in the human body? The precise answer to this question will lay a solid basis for determining how potent the inhibitors are really required and what dosage regimen (dose, frequency, duration, cycle, route, etc) is proper in clinic. (2) How to prevent, diminish and/or circumvent tumor resistance to PARP inhibitors? In fact, PARP inhibitors have been reported to cause drug resistance via secondary mutations in BRCA-deficient cancers63, 64. Therefore, it is necessary to intensify the study on the drug resistance, in particular, on its molecular mechanisms, its relevance to the clinical dosage regimens, its preventive measures and the approach to overcoming it. (3) What are the proper biomarkers for surveillance over therapy, drug resistance and toxicity? Because PARP inhibitors elicit anticancer effects in an indirect manner (ie required for the involvement of the HR pathway), only using the enzymatic activity of the PARP(s) as the biomarker is not enough. Specific defects of the HR pathway and the DSB signaling pathway (for example, BRCA1/2, ATM, ChK1, PTEN, Rad51, etc) may also be considered. More efforts are needed to establish accurate links between the potential biomarkers and the therapeutic responses, drug resistance and toxicity. (4) How to expand the clinical indications? Due to only small population of patients with cancers carrying BRCA mutations, either genetic or sporadic65, researchers are endeavoring to search for gene mutations related to malignancies, including ATM, Rad51, Chk1, and PTEN29, 61, 66, 67 in hopes to expand the potential clinical application of PARP inhibitors. Except for DNA damaging agents, new types of anticancer agents such as certain molecular-targeted drugs could also come into the arsenal of drug combination for PARP inhibitors. (5) How is the long-term toxicity of PARP inhibitors? Though only low short-term toxicity is observed when the inhibitors are used as a single agent, the long-term toxicity remains to be clarified in clinic. The answer to this question is practically critical in determining whether and how to use the inhibitors, especially in young patients. (6) How to balance the benefit and toxicity risk when PARP inhibitors are used in combination? The current clinical data show that it is almost unavoidable to increase the systematic toxicity risk of DNA damaging agents when used in combination with PARP inhibitors. So carefully choosing the drugs and making rational drug administration regimens are necessary. (7) Whether is the administration schedule of a PARP inhibitor related to its toxicity? Clinical results revealed that the oral PARP inhibitor AZD2281 when dosed continuously caused dose-limiting myelosuppression whereas the intravenous PARP inhibitor BSI-201 did not when given in an intermittent twice weekly schedule though both were in combination with platinum drugs and gemcitabine31. So the data appear to suggest that the drug administration regimens (including the route, interval, duration, etc) of PARP inhibitors may be relevant to their systematic toxicity. (8) How to avoid the off-target effects? In addition to PARPs, ADP-ribose transferase (ARTs), cADPR-ribose synthases (CD38 and CD157) and sirtunins all consume NAD+ as a substrate68. The competitive PARP inhibitors designed to mimic nicotinamide may also affect the functions of these enzymes that would be required to be clarified.
Concluding remarks
Undoubtedly, the study on PARP inhibitors is one of the fastest moving and most exciting fields in research and development of new anticancer drugs in recent years. This would provide one of the most promising new therapeutic approaches to cancers, particularly cancers with specific defects. Nevertheless, as already discussed, there still are many questions and problems in this area. Whether and how to answer the questions and to tackle the problems may determine the future of PARP inhibitors as cancer therapeutics. The ongoing phase III clinical trials with AZD2281 and BSI-201 could partially solve them, but considerable efforts will be definitely required for the final application of PARP inhibitors in clinic.
Abbreviations
- PARP:
-
poly(ADP-ribose) polymerase
- PAR:
-
poly(ADP-ribose)
- MAR:
-
mono(ADP-ribose)
- NAD+:
-
nicotinamide adenine dinucleotide
- Tank:
-
tankyrase
- HR:
-
homologous recombination
- BER:
-
base excision repair
- SSB:
-
DNA single strand breaks
- DSB:
-
DNA double strand breaks
- TNBCs:
-
triple-negative metastatic breast cancers
- APC:
-
adenomatous polyposis coli
- GSK3α/β:
-
glycogen synthase kinase 3α/β
- CKIα:
-
casein kinase I alpha
- Axin:
-
axis inhibitor
- BRCA1:
-
breast cancer type 1 susceptibility protein
- BRCA2:
-
breast cancer type 2 susceptibility protein
- XRCC1:
-
X-ray repair complementing defective repair in Chinese hamster cells 1
- CENP-A:
-
centromere protein A
- CENP-B:
-
centromere protein B
- BUB3:
-
budding uninhibited by benzimidazoles 3 homolog (yeast)
- TRF1:
-
telomere repeat-binding factor 1
- ATM:
-
ataxia telangiectasia mutated
- Chk1:
-
checkpoint kinase 1
- PTEN:
-
phosphatase and tensin homolog deleted on chromosome ten
- IC50:
-
half maximal inhibitory concentration
- EGFR:
-
epidermal growth factor receptor
- VEGFR:
-
vascular endothelial growth factor receptor
- PDGFR:
-
platelet derived growth factor receptor.
References
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005; 434: 913–7.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–21.
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG . PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010; 10: 293–301.
Schreiber V, Dantzer F, Ame JC, de Murcia G . Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 2006; 7: 517–28.
Ferraris DV . Evolution of Poly(ADP-ribose) Polymerase-1 (PARP-1) Inhibitors. From Concept to Clinic. J Med Chem 2010; 53: 4561–84.
Chambon P, Weill JD, Mandel P . Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun 1963; 11: 39–43.
Kleine H, Poreba E, Lesniewicz K, Hassa PO, Hottiger MO, Litchfield DW, et al. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol Cell 2008; 32: 57–69.
Loseva O, Jemth AS, Bryant HE, Schuler H, Lehtio L, Karlberg T, et al. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J Biol Chem 2010; 285: 8054–60.
Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem 1999; 274: 17860–8.
Johansson M . A human poly(ADP-ribose) polymerase gene family (ADPRTL): cDNA cloning of two novel poly(ADP-ribose) polymerase homologues. Genomics 1999; 57: 442–5.
Saxena A, Saffery R, Wong LH, Kalitsis P, Choo KH . Centromere proteins Cenpa, Cenpb, and Bub3 interact with poly(ADP-ribose) polymerase-1 protein and are poly(ADP-ribosyl)ated. J Biol Chem 2002; 277: 26921–6.
Saxena A, Wong LH, Kalitsis P, Earle E, Shaffer LG, Choo KH . Poly(ADP-ribose) polymerase 2 localizes to mammalian active centromeres and interacts with PARP-1, Cenpa, Cenpb and Bub3, but not Cenpc. Hum Mol Genet 2002; 11: 2319–29.
El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW . A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res 2003; 31: 5526–33.
Ding J, Miao ZH, Meng LH, Geng MY . Emerging cancer therapeutic opportunities target DNA-repair systems. Trends Pharmacol Sci 2006; 27: 338–44.
Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem 2002; 277: 23028–36.
Smith S, de Lange T . Tankyrase promotes telomere elongation in human cells. Curr Biol 2000; 10: 1299–302.
Kaminker PG, Kim SH, Taylor RD, Zebarjadian Y, Funk WD, Morin GB, et al. TANK2, a new TRF1-associated poly(ADP-ribose) polymerase, causes rapid induction of cell death upon overexpression. J Biol Chem 2001; 276: 35891–9.
Dynek JN, Smith S . Resolution of sister telomere association is required for progression through mitosis. Science 2004; 304: 97–100.
Chiang YJ, Hsiao SJ, Yver D, Cushman SW, Tessarollo L, Smith S, et al. Tankyrase 1 and tankyrase 2 are essential but redundant for mouse embryonic development. PLoS One 2008; 3: e2639.
Brody LC . Treating cancer by targeting a weakness. N Engl J Med 2005; 353: 949–50.
Yelamos J, Schreiber V, Dantzer F . Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol Med 2008; 14: 169–78.
Ball LG, Xiao W . Molecular basis of ataxia telangiectasia and related diseases. Acta Pharmacol Sin 2005; 26: 897–907.
Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, et al. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair (Amst) 2004; 3: 1389–407.
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA . DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8: 193–204.
Welcsh PL, King MC . BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum Mol Genet 2001; 10: 705–13.
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE . Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343: 692–5.
Cancer risks in BRCA2 mutation carriers. The Breast Cancer Linkage Consortium. J Natl Cancer Inst 1999; 91: 1310–6.
Rosen EM, Fan S, Goldberg ID . BRCA1 and prostate cancer. Cancer Invest 2001; 19: 396–412.
Williamson CT, Muzik H, Turhan AG, Zamo A, O'Connor MJ, Bebb DG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther 2010; 9: 347–57.
Zhu Z . Targeted cancer therapies based on antibodies directed against epidermal growth factor receptor: status and perspectives. Acta Pharmacol Sin 2007; 28: 1476–93.
Drew Y, Plummer R . PARP inhibitors in cancer therapy: two modes of attack on the cancer cell widening the clinical applications. Drug Resist Updat 2009; 12: 153–6.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A 2008; 105: 17079–84.
Ancelin K, Brunori M, Bauwens S, Koering CE, Brun C, Ricoul M, et al. Targeting assay to study the cis functions of human telomeric proteins: evidence for inhibition of telomerase by TRF1 and for activation of telomere degradation by TRF2. Mol Cell Biol 2002; 22: 3474–87.
van Steensel B, de Lange T . Control of telomere length by the human telomeric protein TRF1. Nature 1997; 385: 740–3.
Chang W, Dynek JN, Smith S . TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev 2003; 17: 1328–33.
Cook BD, Dynek JN, Chang W, Shostak G, Smith S . Role for the related poly(ADP-Ribose) polymerases tankyrase 1 and 2 at human telomeres. Mol Cell Biol 2002; 22: 332–42.
Donigian JR, de Lange T . The role of the poly(ADP-ribose) polymerase tankyrase1 in telomere length control by the TRF1 component of the shelterin complex. J Biol Chem 2007; 282: 22662–7.
Huang H, He X . Wnt/beta-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol 2008; 20: 119–25.
Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009; 461: 614–20.
Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 2009; 5: 100–7.
Mendeleyev J, Kirsten E, Hakam A, Buki KG, Kun E . Potential chemotherapeutic activity of 4-iodo-3-nitrobenzamide. Metabolic reduction to the 3-nitroso derivative and induction of cell death in tumor cells in culture. Biochem Pharmacol 1995; 50: 705–14.
Szabo C, Virag L, Cuzzocrea S, Scott GS, Hake P, O'Connor MP, et al. Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly(ADP-ribose) synthase. Proc Natl Acad Sci USA 1998; 95: 3867–72.
Bauer PI, Mendeleyeva J, Kirsten E, Comstock JA, Hakam A, Buki KG, et al. Anti-cancer action of 4-iodo-3-nitrobenzamide in combination with buthionine sulfoximine: inactivation of poly(ADP-ribose) polymerase and tumor glycolysis and the appearance of a poly(ADP-ribose) polymerase protease. Biochem Pharmacol 2002; 63: 455–62.
Melisi D, Ossovskaya V, Zhu C, Rosa R, Ling J, Dougherty PM, et al. Oral poly(ADP-ribose) polymerase-1 inhibitor BSI-401 has antitumor activity and synergizes with oxaliplatin against pancreatic cancer, preventing acute neurotoxicity. Clin Cancer Res 2009; 15: 6367–77.
Ruf A, Mennissier de Murcia J, de Murcia G, Schulz GE . Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc Natl Acad Sci USA 1996; 93: 7481–5.
Oliver AW, Ame JC, Roe SM, Good V, de Murcia G, Pearl LH . Crystal structure of the catalytic fragment of murine poly(ADP-ribose) polymerase-2. Nucleic Acids Res 2004; 32: 456–64.
Papeo G, Forte B, Orsini P, Perrera C, Posteri H, Scolaro A, et al. Poly(ADP-ribose) polymerase inhibition in cancer therapy: are we close to maturity? Expert Opin Ther Pat 2009; 19: 1377–400.
Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Soto M, et al. Poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors in cancer chemotherapy. Recent Pat Anticancer Drug Discov 2006; 1: 39–53.
Lehtio L, Jemth AS, Collins R, Loseva O, Johansson A, Markova N, et al. Structural basis for inhibitor specificity in human poly(ADP-ribose) polymerase-3. J Med Chem 2009; 52: 3108–11.
Moroni F, Formentini L, Gerace E, Camaioni E, Pellegrini-Giampietro DE, Chiarugi A, et al. Selective PARP-2 inhibitors increase apoptosis in hippocampal slices but protect cortical cells in models of post-ischaemic brain damage. Br J Pharmacol 2009; 157: 854–62.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361: 123–34.
Hay T, Matthews JR, Pietzka L, Lau A, Cranston A, Nygren AO, et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res 2009; 69: 3850–5.
O'Shaughnessy J, Osborne C, Pippen J, Yoffe M, Patt D, Monaghan G, et al. Efficacy of BSI-201, a poly (ADP-ribose) polymerase-1 (PARP1) inhibitor, in combination with gemcitabine/carboplatin (G/C) in patients with metastatic triple-negative breast cancer (TNBC): results of a randomized phase II trial. J Clin Oncol 2009; 27: Abstr 3.
Sanofi-aventis. http://en.sanofi-aventis.com/binaries/20090531_bsi_201_en_en_tcm28-25235.pdf2009.
O'Shaughnessy J, Yoffe M, Osborne C, Blum J, Rocha C, Ossovskaya V, et al. Triple negative breast cancer: a phase 2, multi-center, open-label, randomized trial of gemcitabine/carboplatin (G/C), with or without BSI-201, a PARP inhibitor. San Antonio Breast Cancer Symposium Annual Meeting, December 2008.
O'Shaughnessy J, Osborne C, Pippen J, Patt D, Rocha C, Ossovskaya V, et al. Final Results of a Randomized Phase II Study Demonstrating Efficacy and Safety of BSI-201, a Poly (ADP-Ribose) Polymerase (PARP) Inhibitor, in Combination with Gemcitabine/Carboplatin (G/C) in Metastatic Triple Negative Breast Cancer (TNBC). Cancer Res 2009; 69 (24 Suppl): Abstract nr 3122.
Isakoff SJ . Triple-negative breast cancer: role of specific chemotherapy agents. Cancer J 2010; 16: 53–61.
Venkitaraman R . Triple-negative/basal-like breast cancer: clinical, pathologic and molecular features. Expert Rev Anticancer Ther 2010; 10: 199–207.
Robert I, Dantzer F, Reina-San-Martin B . Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J Exp Med 2009; 206: 1047–56.
Yelamos J, Monreal Y, Saenz L, Aguado E, Schreiber V, Mota R, et al. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J 2006; 25: 4350–60.
AstraZenecka. WO2008146035 2008.
Rodon J, Iniesta MD, Papadopoulos K . Development of PARP inhibitors in oncology. Expert Opin Investig Drugs 2009; 18: 31–43.
Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T . Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 2008; 68: 2581–6.
Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008; 451: 1116–20.
Tuma RS . PARP inhibitors: will the new class of drugs match the hype? J Natl Cancer Inst 2009; 101: 1230–2.
Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 2009; 1: 315–22.
McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006; 66: 8109–15.
Penberthy WT, Tsunoda I . The importance of NAD in multiple sclerosis. Curr Pharm Des 2009; 15: 64–99.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (NSFC) (No 30772588 and No 30721005), the National Science & Technology Major Project“Key New Drug Creation and Manufacturing Program”of China (No 2009ZX09301-001) and the Science and Technology Commission of Shanghai Municipality (STCSM) (No 08DZ1980200).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
He, Jx., Yang, Ch. & Miao, Zh. Poly(ADP-ribose) polymerase inhibitors as promising cancer therapeutics. Acta Pharmacol Sin 31, 1172–1180 (2010). https://doi.org/10.1038/aps.2010.103
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2010.103
Keywords
This article is cited by
-
Glycogen synthase kinase 3β inhibition synergizes with PARP inhibitors through the induction of homologous recombination deficiency in colorectal cancer
Cell Death & Disease (2021)
-
Polymerase independent repression of FoxO1 transcription by sequence-specific PARP1 binding to FoxO1 promoter
Cell Death & Disease (2020)
-
Discovery of potent 2,4-difluoro-linker poly(ADP-ribose) polymerase 1 inhibitors with enhanced water solubility and in vivo anticancer efficacy
Acta Pharmacologica Sinica (2017)
