
Abstract
Poly (ADP-ribose) polymerase inhibitors (PARPi) have significantly changed the treatment landscape for tumours harbouring defects in genes involved in homologous repair (HR) such as BRCA1 and BRCA2. Despite initial responsiveness to PARPi, tumours eventually develop resistance through a variety of mechanisms. Rational combination strategies involving PARPi have been explored and are in various stages of clinical development. PARPi combinations have the potential to enhance efficacy through synergistic activity, and also potentially sensitise innately PARPi-resistant tumours to PARPi. Initial combinations involving PARPi with chemotherapy were hindered by significant overlapping haematologic toxicity, but newer combinations with fewer toxicities and more targeted approaches are undergoing evaluation. In this review, we discuss the mechanisms of PARPi resistance and review the rationale and clinical evidence for various PARPi combinations including combinations with chemotherapy, immunotherapy, and targeted therapies. We also highlight emerging PARPi combinations with promising preclinical evidence.
Similar content being viewed by others


Introduction
Tumorigenesis is characterised by the hallmark of genomic instability resulting in the accumulation of molecular aberrancies that contribute to the development, progression, and resistance of cancer cells. However, the rapid accumulation of DNA damage in cancer cells leads to increased dependency on specific DNA repair pathways to respond to otherwise potentially cytotoxic DNA damage. The most common type of DNA damage are single-stranded breaks (SSBs) whereas their counterpart, double-stranded breaks (DSBs), are the more lethal form of DNA damage. A critical component of SSB repair, poly (ADP-ribose) polymerase (PARP) remodels chromatin to recruit DNA repair complexes and enables the progression of stalled replication forks [1]. On the other hand, DSBs are repaired by two main pathways: error-prone nonhomologous end joining (NHEJ), or the high-fidelity homologous recombination (HR) repair pathway, which utilises replicated sister DNA as a template [2]. The latter relies on several key proteins including BRCA1/BRCA2 and RAD51 among others [3].
The current generation of PARP inhibitors (PARPi) competitively binds the NAD+ binding moiety on PARP1 and 2, which leads to the induction of DSBs which in turn rely on HR for accurate repair. The concept of PARP trapping, whereby PARP inhibition prevents dissociation of PARPs from DNA, preventing the access of repair proteins and increasing DSBs, has been implicated as a mechanism of action [4]. Indeed, it was elucidated that PARP inhibition was fatal in BRCA1 and BRCA2-deficient cells [5, 6]. This discovery served as a proof-of-concept for clinical approaches and ushered in a new era of cancer-directed therapy. Clinical development of PARPis was heralded by the arrival of olaparib, which was the first to obtain regulatory approval in 2014 as a maintenance strategy in patients with recurrent ovarian cancer [7]. Following the approval of olaparib, various other PARPis including rucaparib, niraparib, and talazoparib have since obtained regulatory approval for various indications in patients with ovarian cancer, breast cancer, prostate cancer, and pancreatic cancer [8].
While PARPis have successfully shifted treatment paradigms in tumours harbouring HR deficiency (HRD), most patients eventually experience resistance attributed to the restoration of homologous recombination repair, amongst other diverse mechanisms [9]. Therefore, it is possible that combination strategies co-targeting pathways that contribute to these described mechanisms of PARPi resistance could result in a deeper and more durable response. In this review, we will discuss current evidence surrounding mechanisms of PARPi resistance and rational PARPi combinations that have been proposed, with a focus on strategies currently in clinical development.
Mechanisms of PARPi resistance
While PARPis have improved outcomes for a variety of malignancies harbouring HRD, a significant proportion of HRD tumours are innately resistant to PARPi, and those with initial disease control eventually experience disease progression due to the development of a variety of resistance mechanisms. While the mechanisms of innate resistance are poorly understood, several acquired resistance mechanisms have been described.
Restoration of functional homologous recombination repair
Restoration of at least partial homologous recombination repair function is one of the most well-described mechanisms of resistance that may develop after exposure to platinum therapy or PARP inhibition [10, 11]. Secondary reversion mutations that enable restoration of the HR gene’s open reading frame, allowing for complete transcription of the homologous repair genes, are a well-described mechanism of HR restoration [12]. Reversion mutations have been clinically identified in numerous PARPi-treated tumours and detection in circulating tumour DNA has been associated with treatment resistance or reduced response [12,13,14]. Reversion mutations have been reported in circulating tumour DNA of up to 39% of patients with BRCA1/2-mutated prostate cancer after progression on rucaparib, and approximately 24% of patients with ovarian cancer after treatment with platinum chemotherapy and PARPi [14, 15]. When a BRCA1/2 reversion mutation was identified in pretreatment samples from patients with ovarian cancer receiving rucaparib, the median progression-free survival (PFS) was noted to be shorter compared with those without reversion mutations (1.8 months versus 9 months) [14]. Further studies which longitudinally investigate for the development of HR gene (e.g.: BRCA1/2, RAD51C/D, PALB2) reversion mutations through a sequential tumour or liquid biopsies are required to comprehensively inform the real-world incidence, timing of development, and impact of such mutations on patient outcomes in clinical settings.
Epigenetic silencing of BRCA1 or RAD51C is a mechanism of homologous recombination deficiency in up to 5–30% of breast and ovarian cancers, with silencing of all BRCA copies (homozygous methylation) associated with improved PFS in response to rucaparib compared to BRCA1/2-intact cases [16]. However, acquired demethylation of epigenetically silenced BRCA1 and RAD51C promotors has been shown to restore mRNA expression [16,17,18,19]. Indeed, among patients with BRCA wild-type ovarian cancer with high methylation levels in an archival sample (n = 17), none of the patients with methylation loss at enrollment responded to rucaparib versus 38% of patients with maintained methylation [20]. Methylation loss leading to restoration of functional BRCA1/2 can therefore confer PARPi resistance.
Thirdly, alternative mRNA splicing can induce hypomorphic BRCA isoforms which restore HR function. Cancer cell lines and tumours harbouring mutations in exon 11 of BRCA1 express a BRCA1-Δ11q splice variant lacking the majority of exon 11 but retains partial BRCA activity, mediating PARPi resistance [21]. Similarly, expression of RING domain-deficient BRCA1 (Rdd-BRCA1) has been observed in mammary tumours of mice carrying the BRCA185stop founder mutation which does not require interaction with BARD1 for protein stability, enabling normal HR function in the presence of PARP inhibition [22]. Development of hypomorphic BRCA1 isoforms has been described in patient-derived tumour xenografts (PDXs) from patients with germline BRCA1/2-mutant breast cancer resistant to PARPi [23].
BRCA1/2-independent restoration of HR
In the absence of functional BRCA1/2, other pathways involved in DNA damage response such as those that regulate DNA end resection can be altered in response to PARPi. One of the best-studied mechanisms, inactivating mutations in the TP53BP1 gene encoding the 53BP1 protein has been implicated in maintaining genetic stability in the setting of BRCA1 loss [24]. 53BP1 in conjunction with the Shieldin complex (REV7, SHLD1, SHLD2 and SHLD3) favours NHEJ by counteracting DSB end resection, which creates the DNA substrate required for HR [25]. Therefore, inactivation of the 53BP1-Shieldin pathway in BRCA1-deficient tumours confers PARPi resistance by restoring DNA end resection and homologous repair of DSBs [23, 26]. Loss of 53BP1 and reduced SHLD1/2 expression has been identified in patient-derived tumour models of PARPi-resistant breast cancer [23, 27]. Among PDXs from patients with germline BRCA1/2-mutated breast cancer, 2 of 9 PARPi-resistant models had somatic mutations in TP53BP1 and exposure to olaparib in PDX models correlated with reduced mRNA expression of SHLD1 and SHLD2 [23, 27]. In addition to HR and NHEJ, DSBs can also be repaired by alternative end joining (TMEJ), mediated by DNA Polymerase Theta (Polθ), and is relied upon particularly in the absence of NHEJ factors such as 53BP1 or members of the Shieldin complex [28].
DNA replication fork protection
BRCA1/2 are important for DNA replication fork protection, preventing the degradation of stalled replication forks by DNA nucleases [29]. In the absence of BRCA1/2, MRE11 and MUS81 erode replication fork ends, resulting in fork collapse [30]. In BRCA2-deficient cell lines, loss of MLL3/4 complex protein PTIP was associated with decreased MRE11 association with the chromatin which lead to replication fork stability and resistance to PARPi [31]. Alterations such as loss of the nucleosome remodelling factor CHD4 was also shown to decrease recruitment of MRE11 and was present in resistant BRCA2-mutant cells [30, 32]. An independent pathway involved in fork degradation, the EZH2/MUS81 axis, promotes resistance to rucaparib and cisplatin in BRCA2-deficient ovarian cancer cells when downregulated [33].
Ataxia telangiectasia and Rad3-related (ATR) kinase and downstream activation of CHK1 kinase via phosphorylation leads to inhibition of cell cycle progression and restoration of stalled replication forks [34, 35]. In BRCA1-deficient, PARPi-resistant cells, RAD51 loading to DNA DSBs and stalled replication forks occurred in an ATR-dependent fashion [34]. SLFN11 has been identified as a key protein recruited in replication stress that induces irreversible replication block and enhances sensitivity to DNA-damaging agents; and its loss has been associated with PARPi resistance and increased ATR/CHK1 pathway reliance [36]. Expression of SLFN11 has been associated with improved response to DNA-damaging agents in ovarian cancer and small cell lung cancer, and enhancing expression of SLFN11 is a potential strategy under investigation to sensitise tumours to PARPi and other DNA-damaging agents [37].
Diminishing PARP1 trapping
Alterations in the target of PARPi that reduce binding to PARP1 or alter the function of PARP1 have been uncovered as a mechanism of resistance. PARP inhibition impairs DNA replication by generating PARP-DNA adducts; however, downregulation of PARP1 or alterations in the DNA-binding domains of PARP1 render inhibitors of the PARP enzyme ineffective for PARP trapping [38]. Described mutations in PARP1, such as p.R591C in PARPi-resistant tumour samples, were linked to a diminished PARP1 trapping activity on DNA [38]. Similar to other described resistance mechanisms to PARPi, the incidence of acquired or existing PARP1 mutations in tumours remains poorly defined. The PAR glycohydrolase (PARG) enzyme is a component of PARP1 trapping which counterbalances the activity of PARP1 by catabolizing PAR chains, the product of PARP activity. It was shown in BRCA2-mutated mouse mammary tumours that loss of PARG expression enables PARylation despite PARP inhibition, which can contribute to resistance [39].
PARPi efflux
Increased drug efflux has been identified as a well-described mechanism of resistance to PARPi. Mutations that result in overexpression of ABCB1 increase transcription of the drug efflux pump MDR1 (P-glycoprotein), and were found in tissue samples from PARPi-treated breast and ovarian cancers [40]. Indeed, ABCB1 expression was shown to be correlated with resistance to olaparib and rucaparib in ovarian cancer cell lines [41]. In vitro treatment with P-glycoprotein inhibitors has demonstrated the ability to prevent export of PARPi; however, clinical development of such agents has not been successful, likely in part due to the predominance of other resistance mechanisms as well as the toxicity and lack of specificity of these inhibitors [42].
Rational PARPi combinations under investigation
PARPi combinations are an active area of investigation given their potential to overcome various mechanisms of PARPi resistance. Initial combinations evaluated PARPi in combination with DNA-damaging chemotherapy for synergy, however the development of these combinations has been limited by overlapping toxicity such as myelosuppression. Newer approaches such as antibody-drug conjugates (ADCs) are being evaluated to capitalise on potential synergy with less overlapping toxicity. Other combinations generally fall into a few distinct categories: agents that induce an HRD-like phenotype by inhibiting pathways that promote HR and agents that target alternative DDR pathways which are relied upon during replication stress in response to PARPi. The former includes targeting of commonly upregulated cancer pathways such as PI3K and RAS signalling while the latter includes cell cycle checkpoints such as ATR and WEE1-kinase. Another promising approach, immunotherapy combinations have potential synergistic activity with limited overlapping toxicity.
PARPi and targeted therapies
PARPi and PI3K/AKT/MTOR inhibition
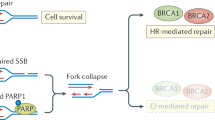
Effective blockade of the PI3K/mTOR pathway has been hypothesised to induce HRD via various mechanisms including suppression of DNA double-strand repair protein SUV39H1 and suppression of homologous recombination repair gene expression [43]. Combinations of PI3K inhibition with PARPi have been investigated in preclinical studies and early-phase clinical trials as an approach to extend the benefit of PARPi to tumours lacking HRD (Fig. 1). In one study, PI3K inhibition in BRCA-proficient triple-negative breast cancer PDX models resulted in ERK activation and downstream activation of ERK-activated transcription factor (ETS1) to downregulate expression of BRCA1/2 [44]. To date, early-phase trials evaluating the combination of olaparib with the pan-PI3K inhibitor buparlisib have demonstrated preliminary efficacy (ORR 30%) in patients with breast and ovarian cancer regardless of BRCA1/2-mutation status, although at the cost of adverse events such as mood disturbance requiring multiple dose reductions of buparlisib [45]. Similarly, the PARPi olaparib has been combined with the α-selective PI3K inhibitor alpelisib in a Phase 1b study of recurrent triple-negative or germline BRCA1 and BRCA2-mutated breast cancer [46]. In this study, 3 of 17 evaluable patients achieved a partial response, none of whom harboured a germline BRCA1/2 mutation [46]. In patients with recurrent and predominantly platinum-resistant ovarian cancer, ORR was 36% with responses seen among those without germline HRR mutations [47]. These early studies appear to indicate that PI3K pathway inhibition confers dysfunctional HR repair, which sensitise tumours to PARPi; and is being further evaluated against chemotherapy in platinum-resistant/refractor ovarian cancer without germline BRCA1/2 mutation in the EPIK-O trial (NCT04729387). Inhibition of the downstream target Akt has been identified as an alternative PI3K pathway target in combination with PARPi. In a Phase 1 study enrolling patients with recurrent endometrial, breast, and ovarian cancer to receive Akt inhibitor capivasertib and olaparib, 6 of 38 evaluable patients (19%) achieved a partial response and there were fewer dose-limiting toxicities [48]. There were also several identified molecular correlates of response including increased activation of DNA damage checkpoints and decreased mTOR activation, while receptor tyrosine kinase activity and RAS-MAPK pathway activity were associated with resistance. The combination was also assessed in a Phase 1 trial enrolling patients with BRCA1/2, HRD-altered, and/or PI3KCA-mutated cancers; 14 (25%) patients achieved a partial response and the combination was safe [49]. The most common adverse events included gastrointestinal toxicities with a grade 3 dose-limiting toxicity of rash noted at higher doses of capivasertib. These studies indicate the feasibility and activity of PARP-PI3K pathway co-targeting, yet further data regarding predictive biomarkers of response and how the addition of PI3K pathway targeting affects acquired resistance mechanisms are still awaited from ongoing studies.

The figure displays various pathways that modulate response to DNA damage and are identified as key targets in combination with PARP inhibitors. The receptor tyrosine kinase cascade mediated by the EGFR, VEGF and c-MET receptors with downstream effectors such as the RAS/MEK and PI3K/AKT pathways are hypothesised to increase homologous repair capacity. Pathways and proteins involved in cell cycle progression such as ATR and WEE1 play an integral role in response to DNA damage in addition to emerging targets USP1 and POLQ. DNA damage mediated by PARP inhibitors is hypothesised to increase T-cell recruitment via activation of the cGAS-STING pathway.
PARPi and antiangiogenic therapies
The vascular endothelial growth factor (VEGF) protein family consists of growth factors that promote angiogenesis in the context of hypoxia. Preclinical studies have demonstrated a link between PARP1 and angiogenesis, with PARP1-knockout in mouse models of skin cancer resulting in decreased HIF-1α and consequently reduced angiogenesis [50]. At the same time, induction of hypoxia via inhibition of VEGF is associated with downregulation BRCA1/2 and RAD51, in BRCA1/2 wild-type ovarian cancer cell lines [51,52,53]. In ovarian cancer cell lines, it was shown that cedinarib-induced hypoxia activated the p130/E2F4 complex that binds to E2F consensus sequences in the promoters of HRR genes to decrease transcription [53]. However, others were unable to replicate the HRR-altering effects of cedinarib in orthotopic ovarian cancer PDXs, despite noting synergy of the combination [52]. Consequently, the rational combination of antiangiogenics with PARPi have been explored in clinical studies, primarily in the setting of ovarian cancer. The Phase 3 PAOLA-1 study evaluated the combination of PARPi and antiangiogenic treatment with bevacizumab as a first-line maintenance strategy after initial platinum-based chemotherapy in advanced ovarian cancer [54]. The trial met its primary endpoint of improved median PFS for olaparib plus bevacizumab versus bevacizumab alone, which has led to the regulatory approval of this combination. Combinations of PARPi and bevacizumab or oral VEGF tyrosine kinase inhibitor, cediranib, has also been explored extensively in recurrent ovarian cancer as a chemotherapy-free alternative in the platinum-sensitive and resistant disease setting [55,56,57,58] (Table 1). The combination of niraparib and bevacizumab was evaluated in patients with platinum-sensitive recurrent ovarian cancer where it demonstrated superior PFS compared to niraparib alone (11.9 months vs 5.5 months) [55]. NIRVANA-1 (NCT05183984) and NIRVANA-R (NCT04734665) are assessing the combination in patients after complete cytoreduction and in patients with prior PARPi exposure, respectively.
While the combination of cediranib with olaparib has demonstrated activity as a chemo-free regimen for recurrent platinum-sensitive ovarian cancer, the recent Phase 3 NRG-GY004 trial in patients with platinum-sensitive ovarian cancer did not demonstrate an improvement in PFS with the combinations of olaparib plus cediranib versus platinum-based chemotherapy [56]. Nevertheless, the subgroup of patients with germline BRCA1/2 mutation appeared to benefit most with a PFS hazard ratio versus chemotherapy of 0.55 (95% CI, 0.32–0.94) for olaparib/cediranib and 0.63 (95% CI, 0.37–1.07) for olaparib in this PARPi-naive population (Table 1) [56]. Several other Phase 2 trials have evaluated the olaparib plus cediranib combination in platinum-resistant ovarian cancer with promising activity observed [57, 59, 60] (Table 1). In the AMBITION study HRD-positive patients with heavily pre-treated platinum-resistant ovarian cancer were randomised to receive either olaparib and cediranib versus olaparib and durvalumab with response rates of (8/16) 50% and (6/14) 43% respectively [59]. In both the BAROCCO and OCTOVA trials, the median progression-free survival for the olaparib plus cediranib combination (dosed continuously) trended towards improvement compared with paclitaxel alone [57, 60]. These benefits were observed irrespective of BRCA1/2 genomic status, or prior antiangiogenic or PARPi use, in the OCTOVA trial [60]. In the EVOLVE study, patients with progression on PARPi for recurrent ovarian cancer received olaparib and cedinarib; in this context, only three objective responses (9%) were noted, with several patients having developed reversion mutations in HR genes as proposed mechanism of resistance [58]. Further Phase 3 ongoing trials evaluating this combination include ICON 9 (NCT03278717) and NRG-GY005 (NCT02502266) in patients with the platinum-resistant disease and in the maintenance setting for platinum-sensitive recurrent ovarian cancer, respectively.
There have been relatively limited studies and successes in non-ovarian cancer pathologies such as pancreatic cancer, where antiangiogenics and PARPi have limited activity in HR proficient disease [61]. Ongoing trials are assessing the combination of various antiangiogenic agents with PARPi in gastric, endometrial (NCT03570437), cervical (NCT04487587), breast (NCT04090567) and prostate cancers (NCT02893917) [62]. The PARPi plus antiangiogenic strategy is promising, but integrating such combinations into treatment paradigms needs further data to clarify their role, including molecular correlates of response and impact on PARPi-resistance mechanisms.
PARPi and RAS/RAF/MEK pathway targeting
Promising preclinical data have demonstrated synergism between MEK inhibition and PARP inhibition in vitro and in vivo [63, 64]. MEK inhibition was shown to decrease expression of components of the homologous recombination DNA repair pathway and decrease HR repair capacity, in part by diminishing the expression of FOXO3a, to enable synergy with PARPi, which has led to the evaluation of MEKi and PARPi combinations in the clinic [63]. In an ongoing trial evaluating MEK inhibitor selumetinib in combination with olaparib in primarily KRAS-mutated tumours, 2 of 14 patients had a confirmed partial response, and the combination was safe and tolerable [65].
PARPi and BET inhibition
The BET bromodomain family of proteins is thought to promote oncogenesis and genes related to homologous repair [66]. Preclinical data demonstrated that inhibition of BET protein BRD4 resulted in a homologous repair-deficient phenotype, which was associated with PARPi sensitivity in vitro and in vivo regardless of BRCA1/2 or RAS/RAF status, suggesting that the combination could have activity outside of HRD-positive tumours. This was mechanistically shown to be mediated by the downregulation of CtIP which is an important component of DSB repair [67]. An early-phase trial assessing the BET inhibitor ZEN-3694 with talazoparib in patients with germline BRCA1/2 wild-type tumours demonstrated preliminary efficacy with ORR of 22% and clinical benefit rate of 35% among 51 evaluable patients, with thrombocytopenia reported as the most common adverse event [68]. An ongoing trial is evaluating the combination in patients with prior PARPi exposure or KRAS-mutated tumours (NCT05327010).
PARPi and c-MET inhibition
c-MET has been shown to be a key mediator of PARP inhibition by phosphorylating PARP1 at Tyr90 which increases enzymatic activity of PARP1 and decreases binding of PARPi [69]. The c-MET inhibitor crizotinib demonstrated synergy with PARP inhibition in preclinical breast, lung and ovarian cancer models [69, 70]. A sequential combination of crizotinib and a PARPi resulted in activation of ATM/CHK2 and inhibition of c-Met pathways, contributing to a decrease in RAD51 levels and induced apoptotic cell death in ovarian cancer cell lines [70]. There are several ongoing trials evaluating the combination of PARPi and c-MET tyrosine kinase inhibitors including crizotinib (NCT04693468) and cabozantinib (NCT03425201, NCT05038839) in advanced solid tumours.
PARPi and EGFR/HER2 targeting
Overexpression of the epidermal growth factor receptor (EGFR) induces tumour growth and is involved in regulating DNA damage response in preclinical studies [71]. Preclinical work in hepatocellular carcinoma and breast cancer reveal that interactions between c-MET and EGFR result in the phosphorylation of PARP1 to mediate resistance [72, 73]. Lapatinib was evaluated in combination with veliparib in a Phase 1 study enrolling patients with BRCA1/2 wild-type metastatic TNBC; of 17 evaluable patients, 4 patients achieved a partial response [74]. The combination of pan-EGFR inhibitor neratinib was synergistic with niraparib in ovarian cancer cell lines as well, leading to the ongoing clinical trial evaluating the combination in patients with platinum-resistant, BRCA1/2 wild-type ovarian cancer (NCT04502602) [75]. Other studies including the combination of niraparib with osimertinib (NCT 03891615), trastuzumab (NCT 03368729), and trastuzumab deruxtecan (NCT04585958) in EGFR-positive lung and HER2-positive cancers are underway.
PARPi and immune checkpoint blockade combinations
Anti-PD(L)1 and -CTLA4 Immune checkpoint blockade has rapidly altered the field of oncology, resulting in multiple approvals across tumour types and indications [76]. Efforts have been made to enhance the effectiveness of immunotherapy and broaden indications for use through various combination strategies, especially for tumour subtypes that are conventionally thought to be immunotherapy-resistant [77]. Mechanistically, the combination of PARP inhibition and immunotherapy has been of interest due to synergistic effects noted in preclinical models [78]. PARPi induces DSBs resulting in generation of double-strand DNA (dsDNA) that, through cyclic guanosine monophosphate–adenosine monophosphate synthase (cGAS) binding, results in activation of the stimulator of interferon genes (STING) pathway [78]. The cGAS/STING pathway upregulates chemokines CCL5 and CXCL10 leading to the recruitment of CD8 T cells responsible for anti-tumour immunity [79, 80]. PARPi was also found to coincide with upregulation of immune checkpoint PDL-1 [80]. In preclinical models, it was shown that PARP inhibition and anti-PD1 therapy was synergistic regardless of BRCA1/2 mutation in colon, breast and ovarian experimental models in vitro and in vivo [78, 81]. While this combination does not directly address mechanisms of PARPi resistance, it is hypothesised to expand the benefit of PARPi beyond tumours harbouring a ‘BRCAness‘ or HRD phenotype, with the benefit of minimal overlapping toxicity from both drug classes.
Promising preclinical data have prompted the evaluation of PARPi and anti-PD(L)1 combinations in the clinic. Phase I trials demonstrated overall safety profile and preliminary efficacy in a population of patients with advanced solid tumours, with no new adverse events with the combination compared with either monotherapy [82, 83]. The MEDIOLA trial is a Phase II basket study assessing the efficacy and safety of anti-PD1 agent durvalumab and olaparib in patients with solid tumours and a germline BRCA1/2 mutation [84, 85]. The treatment consisted of olaparib alone for 4 weeks followed by a combination of olaparib and durvalumab until disease progression. Among patients with platinum-sensitive recurrent ovarian cancer who received at least one line of platinum therapy, ORR was 71.9%, with a total of 7 complete responses and the median PFS was 11.1 months. Among the 34 enrolled patients with HER2-negative breast cancer, ORR was 56% including 1 patient with complete response, and median PFS was 6.7 months [84, 86]. Anaemia, neutropenia, and pancreatitis were the most common grade 3 or 4 adverse events, consistent with previous reports from studies assessing PARPi or immunotherapy.
The TOPACIO/KEYNOTE-162 Phase I/II study evaluated the combination of niraparib and pembrolizumab in patients with recurrent platinum-resistant ovarian cancer irrespective of BRCA1/2-mutation status [87]. Among 60 patients with evaluable response, ORR was 18% with disease control rate of 65%, including three patients with complete response [87]. Notably, the response rate with the combination of niraparib and pembrolizumab was 19% among patients with BRCA1/2 wild-type tumours, which compares favourably to previously reported ORR of less than 10% with either agent as a monotherapy in this population [88, 89]. Overall, these data suggest that the combination of PARPi and anti-PD1 therapy is safe and effective, though further data is needed to delineate which populations benefit most from the combination. There are several ongoing trials assessing the combination in non-breast/ovarian histologies, and others evaluating triplet combinations of PARPi, immune checkpoint blockade and a third targeted agent such as antiangiogenic (NCT04361370) or AKT inhibitors (NCT03772561) [90,91,92,93,94]. Translational studies have attempted to characterise features associated with response to the PARP inhibitor and immune checkpoint inhibitor combination [95, 96]. Among patients with ovarian cancer enrolled in the TOPACIO/KEYNOT-162 study evaluating niraparib and pembrolizumab, tumours from responders typically had features of HRD (mutational signature 3) and increased presence of exhausted CD8 + T cells (immune score) versus non-responders [95]. Biopsies from patients receiving durvalumab and olaparib in the MEDIOLA trial revealed increased STING and IFN-1 pathway activity in responders, and lack of increase was associated with resistance to treatment [96].
PARPi and DNA damage response inhibitors
Upregulation of alternative pathways involved in DNA damage and replication stress are implicated as mechanisms for PARPi resistance. Therapeutic approaches targeting proteins involved in replication stress and DNA repair are therefore being investigated as a potential strategy to overcome PARPi resistance.
PARPi and ATR inhibitors
One area of investigation has been the combination of PARPi with inhibitors of the ATR kinase, a protein that is involved in the regulation of cellular response to stalled and collapsed replication forks through downstream activation of the CHK1/WEE1 axis [97, 98]. Moreover, ATR and CHK1 act to stabilise and protect stalled replication forks by mediating fork remodelling in cooperation with RAD51, ZRANB3, and SMARCAL1. Inhibition of the ATR/CHK1 pathway disrupts cell cycle progression, leading to chromosomal aberrations, mitotic catastrophe, and apoptosis [99]. Promising preclinical data has led to the development of several ATR inhibitors that have undergone clinical investigation [100]. PARPi have been shown to result in early activation of ATR/CHK1 pathway, and the combination of olaparib with ATR inhibitor ceralasertib synergistically suppressed growth in PDX models of BRCA1/2-mutant HGSOC [101]. Importantly, the combination has demonstrated activity in the setting of BRCA1/2 reversion mutations which is a common mechanism of PARPi resistance [34, 101]. In terms of clinical data, a Phase 2 study of ceralasertib and olaparib conducted in patients with recurrent, epithelial ovarian cancer has reported preliminary efficacy in this population [102]. In 14 enrolled patients with the platinum-resistant disease who were PARPi-naive, the majority of which had no somatic or germline alterations in HR genes, the best response was stable disease (SD), which was achieved in 9 of 12 evaluable patients and median PFS was 4.2 months; patients with germline or somatic HR alterations appeared to derive increased benefit from the combination. Most toxicities were limited grade 1 or 2, and the combination was well-tolerated. Confirmed biomarkers for ATR inhibitors or ATR and PARPis are not fully elucidated although multiple potential genomic biomarkers have been described in preclinical data [103,104,105]. In vitro models of prostate cancer with ATM loss were sensitive to ATR inhibition which was enhanced with the addition of PARPi [105]. Co-deletion of RB1 and RNASEH2B was associated with PARPi resistance in prostate cancer cell lines due in part to E2F1-induced BRCA2 expression, however, inhibition of ATR was able to overcome this resistance via disruption of E2F1-induced BRCA2 expression [103]. And while CDK12 loss is not classically sensitive to PARPi, the addition of ATR inhibition sensitised CDK12-deficient cells to PARPi [104]. These data highlight the potential for PARPi combinations to overcome intrinsic resistance to PARPi.
PARPi and WEE-like kinase 1 inhibitors
WEE1 modulates cell cycle progression through the S and G2/M checkpoints via regulation of CDK1 and 2, and inhibition has been shown to promote premature mitotic entry and mitotic catastrophe [106, 107]. Inhibitors of WEE1 have therefore been evaluated as a candidate for combination with PARP inhibition. The combination was shown to be synergistic in PARPi-resistant models across several histologies [108,109,110]. Moreover, the WEE1 inhibitor adavosertib alone or in combination with olaparib was assessed in a Phase II study enrolling patients with recurrent ovarian cancer with previous progression on PARPi treatment, approximately half with a germline or somatic BRCA1/2 mutation [111]. Among the 35 patients receiving combination therapy, ORR was 29% versus 23% for adavosertib alone, and the clinical benefit rate was 89%. Due to the overlapping toxicity profile, there was increased myelosuppression and gastrointestinal toxicity from the combination describe the incidence of high grade toxicity [111]. One approach to improve tolerability while maintaining efficacy has been to sequentially dose DDR inhibitors such as WEE1 inhibitors, which was shown to be feasible in ovarian cancer PDX models and on a recently reported Phase I trial [112, 113]. Only 1 of 13 (8%) of patients required a dose reduction for toxicity and disease control was seen despite prior PARPi exposure [113].
PARPi and CDK4/6 inhibitors
Inhibition of cell cycle progression with cyclin-dependent kinase (CDK) inhibitors have greatly altered the landscape for patients with hormone receptor-positive breast cancer, and rational combination of CDK4/6 inhibitors with PARPis has generated interest due to preclinical data demonstrating synergy in TNBC, ovarian, and neuroendocrine prostate cancer [114,115,116,117]. CDK4/6 inhibition was shown to downregulate homologous recombination, sensitising breast and ovarian cancer cell lines to PARP inhibition to inhibit cell growth, particularly in tumours with high levels of MYC expression [117]. It is hypothesised that the synergistic effects from CDK4/6 inhibition are mediated by inhibition of the MYC pathway, which was shown to regulate expression of BRCA1/2 and RAD51 [117]. In prostate cancer cell lines, the combination was shown to suppress the p-Rb1-E2F1 signalling axis leading to inhibition of E2F1 gene targets such as BRCA1/2 and RAD51 among others [116]. On this basis, PARPi and CDK4/6 inhibitor combinations are currently under evaluation including in MYC amplified tumours (NCT04693468).
PARPi with other novel DDR targets—POLQ inhibitors and USP1 inhibitors
In addition to HR and NHEJ, theta-mediated end joining, also known as microhomology-mediated end joining (MMEJ), is an error-prone escape pathway mediating repair of double-stranded DNA breaks which relies on Polθ (POLQ) [28]. Knockdown of POLQ was shown to be lethal in HRD tumours, due to their increased reliance on the MMEJ pathway [118, 119]. It was demonstrated that inhibition of POLQ inhibition with novobiocin was able to overcome PARPi resistance in cell lines with increased replication fork stability and HR capacity [119]. Though, cell lines with a BRCA1/2 reversion were not sensitive to POLQ inhibition. An orally bioavailable inhibitor, ART4215, is being evaluated in early-phase clinical trials as a monotherapy or in combination with PARPi (NCT04991480) [28]. USP1 is a deubquitinase which has emerged as a promising target as it plays a key role in stabilisation of the replication fork [120]. Particularly in BRCA1-deficient cells, USP1 prevents the recruitment of TLS polymerases to the fork by preventing the accumulation of monoubiquinated Proliferating cell nuclear antigen (PCNA-Ub) at the replication fork. In the absence of USP1, BRCA1-deficient cells (but not BRCA2-deficient cells) are unable to tolerate elevated PCNA-Ub levels which lead to TLS polymerase accumulation at the fork, replication fork instability, and mitotic catastrophe. USP1 inhibition suppressed cell growth among PARPi-resistant BRCA1-deficient cell lines with restored replication fork stability. A novel USP1 inhibitor KSQ-4279 is currently undergoing evaluation alone and in combination with PARPi in advanced cancers (NCT05240898).
PARPi and novel hormonal agents
The androgen receptor has been directly implicated in the expression of DNA repair genes such as DNA-dependent protein kinase catalytic subunit (DNA-PKcs), a protein essential for nonhomologous end joining (NHEJ) [121, 122]. Androgen signalling also regulates the expression of XRCC2 and XRCC3, which are important for HR [122]. It has therefore been hypothesised that co-inhibition of PARP and antiandrogens could induce tumour regression independent of the presence of HR alterations. Several Phase 3 trials have read out on combinations of novel hormonal agents with PARPi, with further trials ongoing (Table 1). In the Phase 3 PROpel trial which enrolled patients with metastatic castration-resistant prostate cancer to receive abiraterone plus olaparib versus abiraterone plus placebo, median imaging-based PFS was longer in the combination group (24.8 vs 16.6 months) irrespective of HR status [123]. On the contrary, early results from the Phase 3 MAGNITUDE study assessing the combination of niraparib and abiraterone in a similar patient population revealed a significant benefit with the combination of niraparib and abiraterone in patients with BRCA1/2-mutated or HRR-mutated cancer (defined as ATM, BRCA1, BRCA2, BRIP1, CDK12, CHEK2, FANCA, HDAC2, PALB2 mutations), but not HRR-negative prostate cancer [124]. Longer-term follow-up, translational research, and results from concomitant trials (Table 1) assessing the combination of novel hormonal agents with PARPi are awaited to clarify the value of the combination in patients with metastatic castration-resistant prostate cancer.
PARPi and chemotherapy
The complementary DNA-damaging effects of PARPi and chemotherapy have provided rationale for exploring their combination (Table 1). While these combinations do not directly address resistance mechanisms to PARPi, they may increase the efficacy and delay the development of resistance compared with PARPi monotherapy. Combinations of PARPis with platinum chemotherapy lead to cross-linking of DNA strands, which prevents DNA replication and transcription, leading to cell cycle arrest, and eventual apoptosis [125]. Yet, significant challenges have been met in the development of PARPi—platinum-based chemotherapy combinations due to overlapping myelosuppression which has limited the feasibility of this strategy. This has led to PARPi being developed as a sequential maintenance strategy after the initial response to platinum-based chemotherapy in advanced ovarian cancer, with several PARPi including olaparib, niraparib, and rucaparib demonstrating feasibility and efficacy over placebo as front-line maintenance therapy [126,127,128]. Combinations of PARPi with non-platinum chemotherapies may remain feasible. Temozolomide, an orally administered DNA-alkylator, has demonstrated enhanced cytotoxic effects when combined with PARPis in preclinical studies, independent of HR [129, 130]. Temozolomide induces base damage which leads to the formation of PARP-DNA complexes which, when combined with potent PARP-trapping agents, enhances cytotoxicity. This has prompted investigation into trials assessing this combination across various tumour histologies including colorectal cancer, small cell lung cancer, breast cancer, glioblastoma, prostate cancer, and melanoma [131,132,133,134,135,136]. Generally, the combination of temozolomide and PARP inhibition was shown to be well-tolerated; however, efficacy was modest in most tumour subtypes (Table 1). Topoisomerase inhibitors such as irinotecan fix Top1 onto DNA and generate unrepaired SSBs which are lethal in malignant cells. PARP1 is critical for repair of Top1 cleavage sites in DNA, thus PARP inhibition results in increased generation of SSBs and synergism in preclinical models [137]. The therapeutic efficacy and safety of PARP inhibition and irinotecan have been explored in clinical studies, but reports of increased haematologic and gastrointestinal toxicities have limited their adoption [138]. Antibody-drug conjugates (ADC) have been developed to deliver cytotoxic chemotherapy in a more targeted fashion to target-expressing cancer cells; these have the advantage of wider therapeutic windows and reduced toxicity [139]. Sacituzumab govitecan, a tumour-associated calcium signal transducer 2 (Trop-2) targeting ADC containing the active metabolite of irinotecan (SN-38) has been assessed in an early-phase trial in combination with the PARPi rucaparib. It has so far demonstrated promising clinical activity including patients with prior exposure to PARPis, with less overlapping toxicity than seen in PARPi/irinotecan combinations [140]. The ongoing PETRA trial is assessing PARPi in combination with trastuzumab deruxtecan and Trop-2-targeting ADC datopotamab (NCT04644068).
Conclusion
PARPis are versatile drugs with established antitumor activity in a number of settings. PARPi resistance is a key challenge that has prompted investigation into numerous rational combinations at various stages of clinical investigation. So far, there are promising clinical responses which have validated preclinical findings for PARPi combination partners across a variety of drug classes. Further work is needed to optimise the therapeutic window of PARPi combinations and identify confirmed predictive biomarkers of response to therapies to refine patient selection and maximise PARPi benefit.
Data availability
Not applicable.
References
Bai P. Biology of poly(ADP-Ribose) polymerases: the factotums of cell maintenance. Mol Cell. 2015;58:947–58.
Ray U, Raghavan SC. Understanding the DNA double-strand break repair and its therapeutic implications. DNA Repair. 2021;106:103177.
Wright WD, Shah SS, Heyer WD. Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem. 2018;293:10524–35.
Murai J, Huang S yin N, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–99.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7.
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382–92.
Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019;30:1437–47.
D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair. 2018;71:172–6.
Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5.
Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20.
Tobalina L, Armenia J, Irving E, O’Connor MJ, Forment JV. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann Oncol. 2021;32:103–12.
Pettitt SJ, Frankum JR, Punta M, Lise S, Alexander J, Chen Y, et al. Clinical BRCA1/2 reversion analysis identifies hotspot mutations and predicted neoantigens associated with therapy resistance. Cancer Discov. 2020;10:1475–88.
Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9:210–9.
Loehr A, Hussain A, Patnaik A, Bryce AH, Castellano D, Font A, et al. Emergence of BRCA reversion mutations in patients with metastatic castration-resistant prostate cancer after treatment with rucaparib. Eur Urol. 2023;83:200–9.
Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun. 2018;9:3970.
Ter Brugge P, Kristel P, van der Burg E, Boon U, de Maaker M, Lips E, et al. Mechanisms of therapy resistance in patient-derived xenograft models of BRCA1-deficient breast cancer. J Natl Cancer Inst. 2016;108.
Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27:1449–55.
Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1gene in sporadic breast cancer. Breast Cancer Res. 2006;8:R38.
Swisher EM, Kwan TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat Commun. 2021;12:2487.
Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016;76:2778–90.
Drost R, Dhillon KK, Gulden H, van der, Heijden I, van der, Brandsma I, Cruz C, et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J Clin Invest. 2016;126:2903–18.
Cruz C, Castroviejo-Bermejo M, Gutiérrez-Enríquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol J Eur Soc Med Oncol. 2018;29:1203–10.
Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010;141:243–54.
Setiaputra D, Durocher D. Shieldin—the protector of DNA ends. EMBO Rep. 2019;20:e47560.
Belotserkovskaya R, Raga Gil E, Lawrence N, Butler R, Clifford G, Wilson MD, et al. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat Commun. 2020;11:819.
Dev H, Chiang TWW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20:954–65.
Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun. 2021;12:3636.
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–42.
Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–7.
Liao H, Ji F, Helleday T, Ying S. Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018;19:e46263.
Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, et al. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015;29:489–94.
Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19:1371–8.
Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31:318–32.
Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16:81–104.
Zhang B, Ramkumar K, Cardnell RJ, Gay CM, Stewart CA, Wang WL, et al. A wake-up call for cancer DNA damage: the role of Schlafen 11 (SLFN11) across multiple cancers. Br J Cancer. 2021;125:1333–40.
Willis SE, Winkler C, Roudier MP, Baird T, Marco-Casanova P, Jones EV, et al. Retrospective analysis of Schlafen11 (SLFN11) to predict the outcomes to therapies affecting the DNA damage response. Br J Cancer. 2021;125:1666–76.
Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9:1849.
Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell. 2018;33:1078–1093.e12.
Christie EL, Pattnaik S, Beach J, Copeland A, Rashoo N, Fereday S, et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat Commun. 2019;10:1295.
Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, Bray SE, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer. 2016;115:431–41.
Lawlor D, Martin P, Busschots S, Thery J, O’Leary JJ, Hennessy BT, et al. PARP inhibitors as P-glyoprotein substrates. J Pharm Sci. 2014;103:1913–20.
Mo W, Liu Q, Lin CCJ, Dai H, Peng Y, Liang Y, et al. mTOR inhibitors suppress homologous recombination repair and synergize with PARP inhibitors via regulating SUV39H1 in BRCA-proficient triple-negative breast cancer. Clin Cancer Res J Am Assoc Cancer Res. 2016;22:1699–712.
Rehman FL, Lord CJ, Ashworth A. The promise of combining inhibition of PI3K and PARP as cancer therapy. Cancer Discov. 2012;2:982–4.
Matulonis UA, Wulf GM, Barry WT, Birrer M, Westin SN, Farooq S, et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann Oncol J Eur Soc Med Oncol. 2017;28:512–8.
Batalini F, Xiong N, Tayob N, Polak M, Eismann J, Cantley LC, et al. Phase 1b clinical trial with alpelisib plus olaparib for patients with advanced triple-negative breast cancer. Clin Cancer Res J Am Assoc Cancer Res. 2022;28:1493–9.
Konstantinopoulos PA, Barry WT, Birrer M, Westin SN, Cadoo KA, Shapiro GI, et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019;20:570–80.
Westin SN, Labrie M, Litton JK, Blucher A, Fang Y, Vellano CP, et al. Phase 1b dose expansion and translational analyses of olaparib in combination with capivasertib in recurrent endometrial, triple negative breast, and ovarian cancer. Clin Cancer Res J Am Assoc Cancer Res. 2021;27:6354–65.
Yap TA, Kristeleit R, Michalarea V, Pettitt SJ, Lim JSJ, Carreira S, et al. Phase I trial of the PARP inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2- and non-BRCA1/2-mutant cancers. Cancer Discov. 2020;10:1528–43.
Martin-Oliva D, Aguilar-Quesada R, O’Valle F, Muñoz-Gámez JA, Martínez-Romero R, García del Moral R, et al. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res. 2006;66:5744–56.
Lim JJ, Yang K, Taylor-Harding B, Wiedemeyer WR, Buckanovich RJ. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia. 2014;16:343–353.e1-2.
Bizzaro F, Fuso Nerini I, Taylor MA, Anastasia A, Russo M, Damia G, et al. VEGF pathway inhibition potentiates PARP inhibitor efficacy in ovarian cancer independent of BRCA status. J Hematol Oncol. 2021;14:186.
Kaplan AR, Gueble SE, Liu Y, Oeck S, Kim H, Yun Z, et al. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci Transl Med. 2019;11:eaav4508.
Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381:2416–28.
Mirza MR, Åvall Lundqvist E, Birrer MJ, de Pont Christensen R, Nyvang GB, Malander S, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol. 2019;20:1409–19.
Liu JF, Brady MF, Matulonis UA, Miller A, Kohn EC, Swisher EM, et al. Olaparib with or without cediranib versus platinum-based chemotherapy in recurrent platinum-sensitive ovarian cancer (NRG-GY004): a randomized, open-label, phase III trial. J Clin Oncol. 2022;40:2138–47.
Colombo N, Tomao F, Benedetti Panici P, Nicoletto MO, Tognon G, Bologna A, et al. Randomized phase II trial of weekly paclitaxel vs. cediranib-olaparib (continuous or intermittent schedule) in platinum-resistant high-grade epithelial ovarian cancer. Gynecol Oncol. 2022;164:505–13.
Lheureux S, Oaknin A, Garg S, Bruce JP, Madariaga A, Dhani NC, et al. EVOLVE: a multicenter open-label single-arm clinical and translational phase II trial of cediranib plus olaparib for ovarian cancer after PARP inhibition progression. Clin Cancer Res J Am Assoc Cancer Res. 2020;26:4206–15.
Lee JY, Kim BG, Kim JW, Lee JB, Park E, Joung JG, et al. Biomarker-guided targeted therapy in platinum-resistant ovarian cancer (AMBITION; KGOG 3045): a multicentre, open-label, five-arm, uncontrolled, umbrella trial. J Gynecol Oncol. 2022;33:e45.
Nicum S, Strauss VY, McGregor N, McNeish I, Roux R, Hall M, et al. OCTOVA: a randomised phase II trial of olaparib, chemotherapy, or olaparib and cediranib in patients with BRCA-mutated platinum-resistant ovarian cancer. Ann Oncol. 2017;28:v352.
Kim JW, Cardin DB, Vaishampayan UN, Kato S, Grossman SR, Glazer PM, et al. Clinical activity and safety of cediranib and olaparib combination in patients with metastatic pancreatic ductal adenocarcinoma without BRCA mutation. Oncologist. 2021;26:e1104–9.
Cecchini M, Chao J, Shyr Y, Cleary J, Uboha N, Cho M, et al. Abstract CT170: a phase 1/2 study of olaparib in combination with ramucirumab in metastatic gastric and gastroesophageal junction adenocarcinoma (GEJ). Cancer Res. 2022;82:CT170.
Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med. 2017;9:eaal5148.
Vena F, Jia R, Esfandiari A, Garcia-Gomez JJ, Rodriguez-Justo M, Ma J, et al. MEK inhibition leads to BRCA2 downregulation and sensitization to DNA damaging agents in pancreas and ovarian cancer models. Oncotarget. 2018;9:11592–603.
Kurnit KC, Meric-Bernstam F, Hess K, Coleman RL, Bhosale P, Savelieva K, et al. Abstract CT020: Phase I dose escalation of olaparib (PARP inhibitor) and selumetinib (MEK Inhibitor) combination in solid tumors with Ras pathway alterations. Cancer Res. 2019;79:CT020.
Mio C, Gerratana L, Bolis M, Caponnetto F, Zanello A, Barbina M, et al. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int J Cancer. 2019;144:755–66.
Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33:401–416.e8.
Aftimos PG, Oliveira M, Punie K, Boni V, Hamilton EP, Gucalp A, et al. A phase 1b/2 study of the BET inhibitor ZEN-3694 in combination with talazoparib for treatment of patients with TNBC without gBRCA1/2 mutations. J Clin Oncol. 2022;40:1023–1023.
Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201.
Sahin ID, Jönsson JM, Hedenfalk I. Crizotinib and PARP inhibitors act synergistically by triggering apoptosis in high-grade serous ovarian cancer. Oncotarget. 2019;10:6981–96.
Li L, Wang H, Yang ES, Arteaga CL, Xia F. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Res. 2008;68:9141–6.
Chu YY, Yam C, Chen MK, Chan LC, Xiao M, Wei YK, et al. Blocking c-Met and EGFR reverses acquired resistance of PARP inhibitors in triple-negative breast cancer. Am J Cancer Res. 2020;10:648–61.
Dong Q, Du Y, Li H, Liu C, Wei Y, Chen MK, et al. EGFR and c-MET cooperate to enhance resistance to PARP inhibitors in hepatocellular carcinoma. Cancer Res. 2019;79:819–29.
Stringer-Reasor EM, May JE, Olariu E, Caterinicchia V, Li Y, Chen D, et al. An open-label, pilot study of veliparib and lapatinib in patients with metastatic, triple-negative breast cancer. Breast Cancer Res. 2021;23:30.
Booth L, Roberts JL, Samuel P, Avogadri-Connors F, Cutler RE, Lalani AS, et al. The irreversible ERBB1/2/4 inhibitor neratinib interacts with the PARP1 inhibitor niraparib to kill ovarian cancer cells. Cancer Biol Ther. 2018;19:525–33.
Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223–49.
Pham MM, Ngoi NYL, Peng G, Tan DSP, Yap TA. Development of poly(ADP-ribose) polymerase inhibitor and immunotherapy combinations: progress, pitfalls, and promises. Trends Cancer. 2021;7:958–70.
Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8+T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019;9:722–37.
Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–14.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res J Am Assoc Cancer Res. 2017;23:3711–20.
Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79:311–9.
Friedlander M, Meniawy T, Markman B, Mileshkin L, Harnett P, Millward M, et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019;20:1306–15.
Lee JM, Cimino-Mathews A, Peer CJ, Zimmer A, Lipkowitz S, Annunziata CM, et al. Safety and clinical activity of the programmed death-ligand 1 inhibitor durvalumab in combination with poly (ADP-ribose) polymerase inhibitor olaparib or vascular endothelial growth factor receptor 1-3 inhibitor cediranib in women’s cancers: a dose-escalation, phase I study. J Clin Oncol. 2017;35:2193–202.
Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020 ;21:1155–64.
Bang YJ, Kaufman B, Geva R, Stemmer SM, Hong SH, Lee JS, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): results in patients with relapsed gastric cancer. J Clin Oncol 2019;37:140–140.
Drew Y, Jonge M, de, Hong SH, Park YH, Wolfer A, Brown J, et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol Oncol. 2018;149:246–7.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5:1141–9.
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–61.
Liu JF, Herold C, Luo W, Penson R, Horowitz N, Konstantinopoulos P, et al. A phase II trial of combination nivolumab and bevacizumab in recurrent ovarian cancer. Ann Oncol. 2018;29:viii334–5.
Wanderley CWS, Correa TS, Scaranti M, Cunha FQ, Barroso-Sousa R. Targeting PARP1 to enhance anticancer checkpoint immunotherapy response: rationale and clinical implications. Front Immunol. 2022;13:816642.
Thomas A, Vilimas R, Trindade C, Erwin-Cohen R, Roper N, Xi L, et al. Durvalumab in combination with olaparib in patients with relapsed SCLC: results from a phase II study. J Thorac Oncol. 2019;14:1447–57.
Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol. 2019;5:1132–40.
Reiss KA, Mick R, Teitelbaum U, O’Hara M, Schneider C, Massa R, et al. Niraparib plus nivolumab or niraparib plus ipilimumab in patients with platinum-sensitive advanced pancreatic cancer: a randomised, phase 1b/2 trial. Lancet Oncol. 2022;23:1009–20.
Yap TA, Bessudo A, Hamilton E, Sachdev J, Patel MR, Rodon J, et al. IOLite: phase 1b trial of doublet/triplet combinations of dostarlimab with niraparib, carboplatin-paclitaxel, with or without bevacizumab in patients with advanced cancer. J Immunother Cancer. 2022;10:e003924.
Färkkilä A, Gulhan DC, Casado J, Jacobson CA, Nguyen H, Kochupurakkal B, et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat Commun. 2020;11:1459.
Staniszewska AD, Armenia J, King M, Michaloglou C, Reddy A, Singh M, et al. PARP inhibition is a modulator of anti-tumor immune response in BRCA-deficient tumors. Oncoimmunology. 2022;11:2083755.
Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res J Am Assoc Cancer Res. 2015;21:4780–5.
Ngoi NYL, Pham MM, Tan DSP, Yap TA. Targeting the replication stress response through synthetic lethal strategies in cancer medicine. Trends Cancer. 2021;7:930–57.
Bartek J, Lukas JChk1. and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9.
Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–30.
Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun. 2020;11:3726.
Shah PD, Wethington SL, Pagan C, Latif N, Tanyi J, Martin LP, et al. Combination ATR and PARP Inhibitor (CAPRI): a phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol. 2021;163:246–53.
Miao C, Tsujino T, Takai T, Gui F, Tsutsumi T, Sztupinszki Z, et al. RB1 loss overrides PARP inhibitor sensitivity driven by RNASEH2B loss in prostate cancer. Sci Adv. 2022;8:eabl9794.
Setton J, Gallo D, Glodzik D, Kaiser B, Braverman S, Ubhi T, et al. CDK12 loss leads to replication stress and sensitivity to combinations of the ATR inhibitor camonsertib (RP-3500) with PARP inhibitors. Eur J Cancer. 2022;174:S99–100.
Neeb A, Herranz N, Arce-Gallego S, Miranda S, Buroni L, Yuan W, et al. Advanced prostate cancer with ATM Loss: PARP and ATR inhibitors. Eur Urol. 2021;79:200–11.
Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–39.
Beck H, Nähse-Kumpf V, Larsen MSY, O’Hanlon KA, Patzke S, Holmberg C, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–36.
Lallo A, Frese KK, Morrow CJ, Sloane R, Gulati S, Schenk MW, et al. The Combination of the PARP inhibitor olaparib and the WEE1 Inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res J Am Assoc Cancer Res. 2018;24:5153–64.
Lin X, Chen D, Zhang C, Zhang X, Li Z, Dong B, et al. Augmented antitumor activity by olaparib plus AZD1775 in gastric cancer through disrupting DNA damage repair pathways and DNA damage checkpoint. J Exp Clin Cancer Res. 2018;37:129.
Ha DH, Min A, Kim S, Jang H, Kim SH, Kim HJ, et al. Antitumor effect of a WEE1 inhibitor and potentiation of olaparib sensitivity by DNA damage response modulation in triple-negative breast cancer. Sci Rep. 2020;10:9930.
Westin SN, Coleman RL, Fellman BM, Yuan Y, Sood AK, Soliman PT, et al. EFFORT: EFFicacy Of adavosertib in parp ResisTance: a randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J Clin Oncol. 2021;39:5505.
Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35:851–67.e7.
Yap TA, Ngoi N, Dumbrava EE, Karp DD, Ahnert JR, Fu S, et al. NCI10329: phase Ib sequential trial of agents against DNA repair (STAR) study to investigate the sequential combination of the poly (ADP-ribose) polymerase inhibitor (PARPi) olaparib (ola) and WEE1 inhibitor (WEE1i) adavosertib (ada) in patients (pts) with DNA damage response (DDR)-aberrant advanced tumors, enriched for BRCA1/2 mutated and CCNE1 amplified cancers. Eur J Cancer. 2022;174:S7.
Konecny GE. Combining PARP and CDK4/6 inhibitors in MYC driven ovarian cancer. EBioMedicine. 2019;43:9–10.
Zhu X, Chen L, Huang B, Li X, Yang L, Hu X, et al. Efficacy and mechanism of the combination of PARP and CDK4/6 inhibitors in the treatment of triple-negative breast cancer. J Exp Clin Cancer Res CR. 2021;40:122.
Wu C, Peng S, Pilie PG, Geng C, Park S, Manyam GC, et al. PARP and CDK4/6 inhibitor combination therapy induces apoptosis and suppresses neuroendocrine differentiation in prostate cancer. Mol Cancer Ther. 2021;20:1680–91.
Yi J, Liu C, Tao Z, Wang M, Jia Y, Sang X, et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine. 2019;43:225–37.
Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518:258–62.
Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat Cancer. 2021;2:598–610.
Suan Lim K, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel L, et al. USP1 is required for replication fork protection in BRCA1-deficient tumors. Mol Cell. 2018;72:925–941.e4.
Asim M, Tarish F, Zecchini HI, Sanjiv K, Gelali E, Massie CE, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun. 2017;8:374.
Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–71.
Clarke NW, Armstrong AJ, Thiery -Vuillemin A, Oya M, Shore N, Loredo E, et al. Abiraterone and olaparib for metastatic castration-resistant prostate cancer. NEJM Evid. 2022;1:EVIDoa2200043.
Chi KN, Rathkopf DE, Smith MR, Efstathiou E, Attard G, Olmos D, et al. Phase 3 MAGNITUDE study: first results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J Clin Oncol. 2022;40:12–12.
Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79.
Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379:2495–505.
González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381:2391–402.
Monk BJ, Parkinson C, Lim MC, O’Malley DM, Oaknin A, Wilson MK, et al. A randomized, phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA–MONO/GOG-3020/ENGOT-ov45). J Clin Oncol. 2022;40:3952–64.
Gill SJ, Travers J, Pshenichnaya I, Kogera FA, Barthorpe S, Mironenko T, et al. Combinations of PARP inhibitors with temozolomide drive PARP1 trapping and apoptosis in Ewing’s sarcoma. PLoS ONE. 2015;10:e0140988.
Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharm Exp Ther. 2014;349:408–16.
Pishvaian MJ, Slack RS, Jiang W, He AR, Hwang JJ, Hankin A, et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer. 2018;124:2337–46.
Farago AF, Yeap BY, Stanzione M, Hung YP, Heist RS, Marcoux JP, et al. Combination olaparib and temozolomide in relapsed small-cell lung cancer. Cancer Discov. 2019;9:1372–87.
Xu J, Keenan TE, Overmoyer B, Tung NM, Gelman RS, Habin K, et al. Phase II trial of veliparib and temozolomide in metastatic breast cancer patients with and without BRCA1/2 mutations. Breast Cancer Res Treat. 2021;189:641–51.
Sim HW, McDonald KL, Lwin Z, Barnes EH, Rosenthal M, Foote MC, et al. A randomized phase II trial of veliparib, radiotherapy, and temozolomide in patients with unmethylated MGMT glioblastoma: the VERTU study. Neuro-Oncol. 2021;23:1736–49.
Hussain M, Carducci MA, Slovin S, Cetnar J, Qian J, McKeegan EM, et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Invest N Drugs. 2014;32:904–12.
Plummer R, Lorigan P, Steven N, Scott L, Middleton MR, Wilson RH, et al. A phase II study of the potent PARP inhibitor, rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharm. 2013;71:1191–9.
Smith LM, Willmore E, Austin CA, Curtin NJ. The novel poly(ADP-Ribose) polymerase inhibitor, AG14361, sensitizes cells to topoisomerase I poisons by increasing the persistence of DNA strand breaks. Clin Cancer Res J Am Assoc Cancer Res. 2005;11:8449–57.
LoRusso PM, Li J, Burger A, Heilbrun LK, Sausville EA, Boerner SA, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the poly(ADP-ribose) polymerase (PARP) inhibitor veliparib (ABT-888) in combination with irinotecan in patients with advanced solid tumors. Clin Cancer Res. 2016;22:3227–37.
Hafeez U, Parakh S, Gan HK, Scott AM. Antibody-drug conjugates for cancer therapy. Molecules. 2020;25:E4764.
Yap TA, Hamilton E, Bauer T, Dumbrava EE, Jeselsohn R, Enke A, et al. Phase Ib SEASTAR study: combining rucaparib and sacituzumab govitecan in patients with cancer with or without mutations in homologous recombination repair genes. JCO Precis Oncol. 2022;6:e2100456.
Ramalingam SS, Novello S, Guclu SZ, Bentsion D, Zvirbule Z, Szilasi M, et al. Veliparib in combination with platinum-based chemotherapy for first-line treatment of advanced squamous cell lung cancer: a randomized, multicenter phase III study. J Clin Oncol. 2021;39:3633–44.
O’Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol. 2020;38:1378–88.
Diéras V, Han HS, Kaufman B, Wildiers H, Friedlander M, Ayoub JP, et al. Veliparib with carboplatin and paclitaxel in BRCA-mutated advanced breast cancer (BROCADE3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:1269–82.
Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, et al. Randomized, double-blind, phase II study of temozolomide in combination with either veliparib or placebo in patients with relapsed-sensitive or refractory small-cell lung cancer. J Clin Oncol. 2018;36:2386–94.
Gorbunova V, Beck JT, Hofheinz RD, Garcia-Alfonso P, Nechaeva M, Cubillo Gracian A, et al. A phase 2 randomised study of veliparib plus FOLFIRI±bevacizumab versus placebo plus FOLFIRI±bevacizumab in metastatic colorectal cancer. Br J Cancer. 2019;120:183–9.
Hardesty MM, Krivak TC, Wright GS, Hamilton E, Fleming EL, Belotte J, et al. OVARIO phase II trial of combination niraparib plus bevacizumab maintenance therapy in advanced ovarian cancer following first-line platinum-based chemotherapy with bevacizumab. Gynecol Oncol. 2022;166:219–29.
Mahdi H, Hafez N, Doroshow D, Sohal D, Keedy V, Do KT, et al. Ceralasertib-mediated ATR inhibition combined with olaparib in advanced cancers harboring DNA damage response and repair alterations (olaparib combinations). JCO Precis Oncol. 2021;5:PO.20.00439.
Do KT, Kochupurakkal B, Kelland S, de Jonge A, Hedglin J, Powers A, et al. Phase 1 combination study of the CHK1 inhibitor prexasertib and the PARP inhibitor olaparib in high-grade serous ovarian cancer and other solid tumors. Clin Cancer Res J Am Assoc Cancer Res. 2021;27:4710–6.
Acknowledgements
The authors do not indicate any acknowledgements.
Funding
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
DB, NN, JH and TY all contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhamidipati, D., Haro-Silerio, J.I., Yap, T.A. et al. PARP inhibitors: enhancing efficacy through rational combinations. Br J Cancer 129, 904–916 (2023). https://doi.org/10.1038/s41416-023-02326-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02326-7
