
Abstract
Aim:
Substantial colocalization of functionally independent α4 nicotinic acetylcholine receptors and 5-HT3 serotonin receptors on presynaptic terminals has been observed in brain. The present study was aimed at addressing whether nicotinic acetylcholine receptors and 5-HT3 serotonin receptors interact on the same presynaptic terminal, suggesting a convergence of cholinergic and serotonergic regulation.
Methods:
Ca2+ responses in individual, isolated nerve endings purified from rat striatum were measured using confocal imaging.
Results:
Application of 500 nmol/L nicotine following sustained stimulation with the highly selective 5-HT3 receptor agonist m-chlorophenylbiguanide at 100 nmol/L resulted in markedly reduced Ca2+ responses (28% of control) in only those striatal nerve endings that originally responded to m-chlorophenylbiguanide. The cross-regulation developed over several minutes. Presynaptic nerve endings that had not responded to m-chlorophenylbiguanide, indicating that 5-HT3 receptors were not present, displayed typical responses to nicotine. Application of m-chlorophenylbiguanide following sustained stimulation with nicotine resulted in partially attenuated Ca2+ responses (49% of control). Application of m-chlorophenylbiguanide following sustained stimulation with m-chlorophenylbiguanide also resulted in a strong attenuation of Ca2+ responses (12% of control), whereas nicotine-induced Ca2+ responses following sustained stimulation with nicotine were not significantly different from control.
Conclusion:
These results indicate that the presynaptic Ca2+ increases evoked by either 5-HT3 receptor or nicotinic acetylcholine receptor activation regulate subsequent responses to 5-HT3 receptor activation, but that only 5-HT3 receptors cross-regulate subsequent nicotinic acetylcholine receptor-mediated responses. The findings suggest a specific interaction between the two receptor systems in the same striatal nerve terminal, likely involving Ca2+-dependent intracellular pathways that regulate these signaling systems at one or more levels.
Similar content being viewed by others


Introduction
Neurotransmitter receptors reside on presynaptic nerve terminals at synapses in brain, where they likely act to regulate neurotransmission. In the striatum, a key component of the psychomotor system1 functioning in reward-related dopaminergic signaling2 that is regulated by nicotine3, presynaptic nicotinic acetylcholine receptors (nAChRs) colocalize with 5-HT3 serotonin receptors as functionally and physically independent pathways4. This suggests convergence of nicotinic cholinergic and serotonergic pathways at the level of the individual presynaptic terminal.
Both presynaptic nAChRs and 5-HT3 receptors induce robust increases in presynaptic Ca2+ on activation4, 5, 6, which, in turn, trigger the release of neurotransmitter7, 8. Increases in [Ca2+]i have also been found to regulate desensitization of the receptors9, 10, possibly involving calcineurin11. Cross-regulation via receptor-mediated increases in [Ca2+]i might be one means by which convergent serotonergic and nicotinic pathways could interact. Cross-talk has been noted for a wide variety of neurotransmitter receptors colocalized on presynaptic terminals12, 13, 14, 15, 16 and this possibility was explored for colocalized nAChRs and 5-HT3 receptors on striatal terminals in the present study.
5-HT3 receptors and nAChRs are members of the Cys-loop ligand-gated ion channel superfamily17, 18. They respond to selective agonist activation by conducting cations, including, most notably, Ca2+19, 20. In striatal terminals, increased [Ca2+]i on activation of nAChRs5 or 5-HT3 receptors6 appear to be mainly the result of Ca2+ entry via the receptor-channels, as the Ca2+ responses were largely insensitive to voltage-gated Ca2+ channel (VGCC) blockade or predepolarization. However, dependence of nicotine-induced dopamine release in striatal terminals on VGCCs has been noted21, 22, 23, and additionally, presynaptic responses mediated by nAChRs in other brain regions, such as hippocampus, display a mixed dependence on VGCCs and Ca2+ influx via the receptor channel22, 24, 25. In any case, the robust increases in [Ca2+]i resulting from presynaptic receptor activation in striatal nerve terminals are relatively sustained, and as such, may trigger activation of Ca2+-dependent intracellular regulators, including CaM-dependent kinase, ERK, and calcineurin26, 27, 28. These intracellular regulators may provide the means for receptor-receptor cross-talk at the level of the presynaptic terminal. Here, we demonstrate cross-regulation between presynaptic 5-HT3 receptors and nAChRs in rat striatal nerve terminals.
Materials and methods
Chemicals Fluo-3/AM was purchased from Molecular Probes (Eugene, OR, USA). The adhesive matrix Cell-Tak was from BD Sciences (Bedford, MA, USA). Percoll was originally from Amersham Pharmacia Biotech AB (Uppsala, Sweden). Ultrapure sucrose was from ICN Biomedicals (Aurora, OH, USA). HEPES (ULTROL grade) was from Calbiochem (San Diego, CA, USA). (–)Nicotine and 1-(m-chlorophenyl)biguanide (mCPBG) were from RBI-Sigma (St Louis, MO, USA). All other chemicals were of the highest reagent grade.
Animals Adult, male Sprague-Dawley rats were obtained from Taconic Farms at 6–8 weeks of age.
Purification of isolated presynaptic nerve terminals Intact isolated presynaptic nerve terminals (synaptosomes) were purified as described previously5. In brief, striata from adult Sprague-Dawley rats were dissected into ice-cold 0.32 mol/L sucrose. The tissue was rapidly homogenized in ice-cold 0.32 mol/L sucrose with a glass-Teflon tissue grinder. Synaptosomes were isolated using the Percoll step gradient method29. The purified synaptosomes were washed with oxygenated HEPES-buffered saline (HBS, pH 7.4) containing 142 mmol/L NaCl, 2.4 mmol/L KCl, 1.2 mmol/L K2HPO4, 1 mmol/L MgCl2, 5 mmol/L D-glucose, and 10 mmol/L HEPES, containing 1 mmol/L Ca2+. The protocol used for this study was approved by the Drexel University College of Medicine Institutional Animal Care and Use Committee.
Measurement of relative Ca2+ levels Fluo-3 was loaded into the purified synaptosomes suspended in HBS containing 1 mmol/L Ca2+, using the acetoxymethyl ester derivatives (AM) of the dye at 5 μmol/L final concentration, for 60 min at 37 °C as previously described5. The dye-loaded synaptosomes were then washed by centrifugation and resuspended in HBS. The preparations were plated onto coverslips coated with Cell-Tak and then inserted into a rapid-exchange Warner perfusion system mounted on a Nikon Diaphot microscope attached to a Nikon PCM 2000 laser-scanning confocal imaging system. Fluorescent images were recorded in response to excitation at 488 nm. During the confocal imaging, the preparations were under constant perfusion at 3–5 mL/min with HBS. Images were typically collected at 4-s intervals, with the first 5 consecutive images collected as a baseline. Each experiment corresponds to sequential images collected using a single preparation subjected to various conditions and/or reagents. Nicotine and mCPBG were used at maximal concentrations5, 6.
Data analysis The quantification of fluorescence intensities associated with individual synaptosomes recorded in digitized images was calculated quantified using OPTIMAS image analysis software (Optimas Co) and then corrected for photobleaching (typically <3%, based on baseline images). Response to depolarization evoked by elevated extracellular K+ was used as a criterion for synaptosomal viability for each preparation. Data are presented as normalized responses (F/F0, where F0 is the fluorescence intensity associated with a given structure at t0). Comparisons were made for peak responses. All experiments were independently replicated at least 3 times. All averaged data are from pooled experiments and are presented as means±SEM. Statistical comparisons of averaged peak amplitudes were made by Student's t-test, with P<0.05 as minimal for significant difference.
Results
Immunostaining of individual isolated terminals from rat striatum revealed that over 90% of the terminals expressing α4 containing nAChRs also express 5-HT3 receptors, whereas terminals expressing non-α4 containing nAChRs displayed no overlap in immunostaining for 5-HT3 receptors4. The colocalized nAChRs and 5-HT3 receptors were shown to be functionally independent on the nerve terminals via pharmacological characterization of agonist-induced Ca2+ responses4. However, the possibility remained that sustained activation of one receptor could affect signaling by the other receptor. Following a positive test stimulation with 100 nmol/L mCPBG, prolonged incubation of striatal synaptosomes with mCPBG (40 min) resulted in substantial attenuation of Ca2+ responses evoked subsequently with 500 nmol/L nicotine (Figure 1), when compared to control nicotine-induced Ca2+ responses evoked following the same long incubation but without mCPBG (28%±3% of control; P<0.05). The regulation of nAChR-mediated Ca2+ responses following 5-HT3 receptor activation developed by 1 min, but increased over tens of minutes (Figure 2). For the subset of synaptosomes that did not initially respond to mCPBG, prolonged incubation with mCPBG had no effect on subsequent stimulation by nicotine (Figure 1, lower traces). The presence of this subset of synaptosomes is consistent with previous findings that <50% of striatal terminals responding to nicotine with increased [Ca2+]i also responded to subsequent stimulation with the 5-HT3 agonist mCPBG4.

Cross-regulation of presynaptic nAChRs on individual isolated terminals from rat striatum by colocalized 5-HT3 receptors. Successive stimulation of striatal synaptosomes with the highly selective 5-HT3 receptor agonist mCPBG at 100 nmol/L followed by stimulation with 500 nmol/L nicotine after an intervening incubation with 100 nmol/L mCPBG. Ca2+ responses in two individual synaptosomes measured using confocal imaging are shown in the sequences of traces at top, selected as representative of responders and non-responders for mCPBG stimulation which subsequently responded to nicotine stimulation4. Averaged initial responses to mCPBG (n=71) and subsequent responses to nicotine in synaptosomes first responding to mCPBG (mCPBG responders; n=71) or not responding to mCPBG (mCPBG non-responders (n=24) are displayed as means±SEM. Relative [Ca2+]i is expressed as F/F0, where F0 represents the fluorescent intensity of the individual synaptosome at t0.

Time-course of cross-regulation of presynaptic nAChRs by colocalized 5-HT3 receptors in isolated terminals from rat striatum. Data are expressed as the percent inhibition of averaged nicotine-induced peak Ca2+ responses (Nic) in striatal synaptosomes that had originally responding to 100 nmol/L mCPBG relative to the averaged nicotine-induced Ca2+ responses in those synaptosomes that did not respond to mCPBG, which are equivalent to control nicotine-induced responses. % Inhibition=[100–(Nic responses/control Nic responses ×100)]. n=2.
Likewise, following a test stimulation with nicotine, prolonged incubation of the striatal terminals with nicotine (40min) resulted in attenuation of mCPBG-evoked Ca2+ responses (Figure 3) only in those terminals initially responding to nicotine. However, the nicotine-induced attenuation of subsequent mCPBG-evoked responses was only partial (49%±6% of control; P<0.05) in comparison to the mCPBG-induced attenuation of subsequent nicotine-evoked responses, and the responses had rather slow kinetics.

Cross-regulation of presynaptic 5-HT3 receptors on individual isolated terminals from rat striatum by colocalized nAChRs. Successive stimulation of striatal synaptosomes with 500 nmol/L nicotine followed by stimulation with 100 nmol/L mCPBG after an intervening incubation with 500 nmol/L nicotine. Ca2+ responses in an individual synaptosome measured using confocal imaging are shown in the sequences of traces at top. Averaged responses to nicotine (n=33) and subsequent responses to mCPBG (n=33) are displayed as means±SEM. Averaged control responses to 100 nmol/L mCPBG (n=50) performed under the same conditions are shown for comparison. Only a subset (about 50%) of striatal synaptosomes that respond to nicotine also respond to mCPBG4. Relative [Ca2+]i is expressed as F/F0, where F0 represents the fluorescent intensity of the individual synaptosome at t0.
Stimulation of presynaptic nAChRs in isolated terminals from striatum by 500 nmol/L nicotine results in inactivation after several mins5, whereas stimulation of striatal presynaptic 5-HT3 receptors by mCPBG leads to little apparent inactivation after tens of minutues6. For comparison, homologous regulation of the receptors was thus examined. Successive stimulation with 100 nmol/L mCPBG resulted in very strong attenuation of the second Ca2+ response (12%±5% of control; P<0.05) after a short intervening wash to remove agonist (Figure 4A). In contrast, no significant attenuation was observed with successive stimulation with nicotine (Figure 4B).

Presynaptic 5-HT3 receptors on isolated terminals from rat striatum display sustained desensitization, whereas presynapic nAChRs do not. Averaged Ca2+ responses to successive stimulation of striatal synaptosomes with 100 nmol/L mCPBG (A; n=4) or with 500 nmol/L nicotine (B; n=8) are shown as means±SEM. A brief (several mins) intervening wash with HBS was performed between stimulations. The sustained desensitization of the 5-HT3 receptor-mediated responses required the presence of external Ca2+ (not shown). Relative [Ca2+]i is expressed as F/F0, where F0 represents the fluorescent intensity of the individual synaptosome at t0.
Discussion
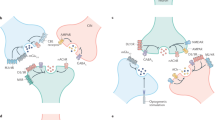
Prolonged activation of presynaptic 5-HT3 receptors on isolated striatal nerve terminals was found to cross-regulate presynaptic nAChRs colocalized on the same terminal (Figure 5). This occurred even though successive stimulation with nicotine did not result in homologous desensitization of presynaptic Ca2+ responses, whereas successive stimulation with the 5-HT3 receptor agonist did. Prolonged stimulation (min) of presynaptic 5-HT3 receptors will result in a sustained increase in [Ca2+]i, owing to the very slow inactivation of this receptor when localized to presynaptic nerve terminals6, which may activate any number of intracellular Ca2+-dependent pathways. In contrast, stimulation of presynaptic nAChRs will result in inactivation5, though the time course for inactivation is still relatively slow when compared to postsynaptic responses30 and some degree of cross-regulation of colocalized 5-HT3 receptor was observed. The latter observation is consistent with an extended incubation with nicotine being necessary for inducing desensitization5, 31.

Cartoon of cross-regulation of presynaptic nAChRs by colocalized 5-HT3 receptors. Prolonged activation of presynaptic 5-HT3 receptors (left) evokes sustained elevations in [Ca2+]i within the nerve terminal. Activation of colocalized presynaptic nAChRs (right) after prolonged activation of presynaptic 5-HT3 receptors results in strongly attenuated Ca2+ responses as compared to control responses in the absence of pretreatment with nAChR agonist. It is postulated that the sustained increase in presynaptic [Ca2+]i on 5-HT3 receptor stimulation activates Ca2+-dependent signaling pathways in the nerve terminal that regulate presynaptic nAChRs, effectively resulting in desensitization.
Cross-talk in presynaptic nerve terminals has been observed for a number of different receptors in conjunction with nAChRs12, 13, 15, 16. Of particular note is the interaction between ionotropic P2X nucleotidic receptors and nAChRs on cholinergic terminals from rat midbrain, which was found to depend on CaM kinase II32. Activation of CaM kinase II and/or calcineurin by sustained increases in presynaptic [Ca2+]i by 5-HT3 receptors, due to their significant Ca2+ permeability20, is an attractive possibility to explain mediation of cross-talk in the present study. However, a number of other intracellular regulators activated by Ca2+ may be candidates for mediating cross-talk between presynaptic 5-HT3 receptors and nAChRs, including ERK and a direct action of CaM. In addition, long-term desensitization of muscle and Torpedo nAChRs, as opposed to rapid inactivation, was found to be regulated by protein kinase A33, 34. Increased Ca2+ could activate protein kinase A via stimulation of Ca2+-regulated adenylyl cyclase35. Finally, direct interaction between colocalized receptors has been proposed for some receptor interactions; however, as the 5-HT3 receptors and nAChRs function independently on the presynaptic terminals4, this is an unlikely mechanism for cross-talk.
Striatal nerve terminals express several subtypes of α4 and α6 containing nAChRs36, 37, including α4β2, α4α5β2, α2α4β2, α4α6β2, and α4α6β3β2. The α4 nAChR subunit was found to colocalize with 5-HT3 receptors on these terminals, but not α54. As 5-HT3 receptors have been linked to the regulation of striatal dopamine release38, 39, colocalization with α2 containing nAChRs is ruled out, because they are only expressed on nondopaminergic structures in the striatum36. The α6 containing nAChRs on striatal terminals appear to display the highest relative affinity for nicotine37, and evoked Ca2+ responses in isolated striatal terminals was observed with relatively low concentrations (50–500 nmol/L) of nicotine5. Taken together, these observations would indicate the likelihood that the colocalized α4 containing nAChRs also contain α6. This postulate remains to be demonstrated. In addition, in view of the relatively slow apparent inactivation of striatal presynaptic nAChRs (min) compared to postsynaptic nAChRs, as noted previously, it would also be of interest to determine which β subunit is present in the receptor, as the particular β subunit will influence the rate of inactivation40.
The functional consequences of cross-regulation between colocalized neurotransmitter receptors have yet to be elucidated. Where a negative effect occurs, here desensitization, it may be that sustained activation of one pathway may be acting to suppress the activity via the other pathway. Thus, intermittent or low frequency stimulation via both pathways may synergize, whereas high frequency, sustained stimulation of one pathway will dominate. As each pathway likely has characteristic patterns of activity, those patterns would be more efficiently transduced to downstream signals follow cross-regulation. Functional interaction of serotonergic and cholinergic signaling in the striatum has been described41. As cholinergic tone plays a key role in regulating the striatal GABAergic output in motor control, mainly via muscarinic receptors, it would be of interest to understand the role that regulation of nAChRs by serotonergic activity plays at dopaminergic presynaptic terminals in psychomotor output from the striatum.
Author contribution
John DOUGHERTY carried out all of the experiments and analyzed the data; Robert NICHOLS designed and supervised all of the experiments and analysis, and wrote the paper.
Abbreviations
HBS, HEPES-buffered saline; VGCC, voltage-gated calcium channel; nAChRs, nicotinic acetylcholine receptors; mCPBG, 1-(m-chlorophenyl)biguanide
References
Hyman SE, Malenka RC . Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2001; 2: 695–703.
Schultz W . Predictive reward signal of dopamine neurons. J Neurophysiol 1998; 80: 1–27.
Rice ME, Cragg SJ . Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci 2004; 7: 583–4.
Nayak SV, Rondé P, Spier AD, Lummis SCR, Nichols RA . Nicotinic receptors co-localize with 5-HT3 serotonin receptors on striatal nerve terminals. Neuropharmacology 2000; 39: 2681–90.
Nayak SV, Dougherty JJ, McIntosh JM, Nichols RA . Ca2+ changes induced by different presynaptic nicotinic receptors in separate populations of individual striatal nerve terminals. J Neurochem 2001; 76: 1860–70.
Rondé P, Nichols RA . High calcium permeability of serotonin 5-HT3 receptors on presynaptic nerve terminals from rat striatum. J Neurochem 1998; 70: 1094–103.
Barik J, Wonnacott S . Molecular and cellular mechanisms of action of nicotine in the CNS. In: Henningfield JE, et al., editors. Handbook of experimental pharmacology, v 192. Nicotinic pharmacology. Heidelberg: Springer-Verlag; 2009. p 173–207.
Barnes NM, Sharp T . A review of central 5-HT receptors and their function. Neuropharmacol 1999; 38: 1083–152.
Jones S, Yakel JL . Ca2+ influx through voltage-gated Ca2+ channels regulates 5-HT3 receptor channel desensitization in rat glioma X mouse neuroblastoma hybrid NG108–15 cells. J Physiol 1998; 510: 361–70.
Khiroug L, Giniatullin R, Klein RC, Fayuk D, Yakel JL . Functional mapping and Ca2+ regulation of nicotinic acetylcholine receptor channels in rat hippocampal CA1 neurons. J Neurosci 2003; 23: 9024–31.
Yakel JL . Calcineurin regulation of synaptic function: from ion channels to transmitter release and gene transcription. Trends Pharmacol Sci 1997; 18: 124–34.
Cheramy A, Godeheu G, L'Hirondel M, Glowinski J . Cooperative contributions of cholinergic and NMDA receptors in the presynaptic control of dopamine release from synaptosomes of the rat striatum. J Pharmacol Exp Ther 1996; 276: 616–25.
Diaz-Hernandez M, Sanchez-Nogueiro J, Pintor J, Miras-Portugal MT . Interaction between dinucleotide and nicotinic receptors in individual cholinergic terminals. J Pharmacol Exp Ther 2004; 311: 954–67.
Rodrigues RJ, Alfaro TM, Rebola N, Oliveira CR, Cunha RA . Colocalization and function interaction between A2A and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J Neurochem 2005: 92: 433–41.
Grilli M, Patti L, Robino F, Zappettini S, Raiteri M, Marchi M . Release-enhancing pre-synaptic muscarinic and nicotinic receptors co-exist and interact on dopaminergic nerve endings of rat nucleus accumbens. J Neurochem 2008; 105: 2205–13.
Grilli M, Zappettini S, Zoli M, Marchi M . Presynaptic nicotinic and D2 receptors functionally interact on dopaminergic nerve endings of rat and mouse nucleus accumbens. J Neurochem 2009; 108: 1507–14.
Ortells MO, Lunt GG . Evolutionary history of the ligand-gated ion-channel superfamily of receptors. Trends Neurosci 1995; 18: 121–7.
Connolly CN, Wafford KA . The Cys-loop superfamily of ligand-gate ion channels: the impact of receptor structure on function. Biochem Soc Trans 2004; 32: 529–34.
Dani JA, Bertrand D . Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol 2007; 47: 699–729.
Peters JA, Hales TG, Lambert JJ . Molecular determinants of single-channel conductance and ion selectivity in the Cys-loop family: insights from the 5-HT3 receptor. Trends Pharmacol Sci 2005; 26: 587–94.
Soliakov L, Wonnacott S . Voltage-sensitive Ca2+ channels involved in nicotinic receptor-medicated [3H]dopamine release from rat striatal synaptosomes. J Neurochem 1996; 67: 163–70.
Kulak JM, McIntosh JM, Yoshikami D, Olivera BM . Nicotine-evoked transmitter release from synaptosomes: functional association of specific presynaptic acetylcholine receptors and voltage-gated calcium channels. J Neurochem 2001; 77: 1581–9.
Turner TJ . Nicotine enhancement of dopamine release by a calcium-dependent increase in the size of the readily releasable pool of synaptic vesicles. J Neurosci 2004; 24: 11328–36.
Girod R, Barazangi N, McGehee D, Role LW . Facilitation of glutamatergic neurotransmission by presynaptic nicotinic acetylcholine receptors. Neuropharmacology 2000; 39: 2715–25.
Dorostkar MM, Boehm S . Opposite effects of presynaptic 5-HT3 receptor activation on spontaneous and action potential-evoked GABA release at hippocampal synapses. J Neurochem 2007; 100: 395–405.
Wang BW, Liao WN, Chang CT, Wang SJ . Facilitation of glutamate release by nicotine involves the activation of a Ca2+/calmodulin signaling pathway in rat prefrontal cortex nerve terminals. Synapse 2006; 59: 491–501.
Hu M, Liu QS, Chang KT, Berg DK . Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci 2002; 21: 616–25.
Liu QS, Berg DK . Actin filaments and the opposing actions of CaM kinase II and calcineurin in regulating α7-containing nicotinic receptors on chick ciliary ganglion neurons. J Neurosci 1999; 19: 10280–8.
Dunkley PR, Jarvie PE, Health JW, Kidd GJ, Rostas JAP . A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res 1986; 327: 115–29.
McGehee DS, Heath MJS, Gelber S, Devay P, Role LW . Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 1995; 269: 1692–6.
Rowell PP, Hillebrand JA . Characterization of nicotine-induced desensitization of evoked dopamine release from rat striatal synaptosomes. J Neurochem 1994; 63: 561–9.
Díaz-Hernandez M, Sánchez-Nogueiro J, Pintor J, Miras-Portugal MT . Interaction between dinucleotide and nicotinic receptors in individual cholinergic terminals. J Pharmacol Exp Ther 2004; 311: 954–67.
Paradiso K, Brehm P . Long-term desensitization of nicotinic acetylcholine receptors is regulated via protein kinase A-mediated phosphorylation. J Neurosci 1998; 18: 9227–37.
Hoffman PW, Ravindran A, Huganir RL . Role of phosphorylation in desensitization of acetylcholine receptors expressed in Xenopus oocytes. J Neurosci 1994; 14: 4185–95.
Willoughby D, Cooper DMF . Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev 2007; 87: 965–1010.
Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C . Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci 2002; 22: 8785–9.
Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 2004; 65: 1526–35.
Blandina P, Goldfarb J, Craddock-Royal B, Green JP . Release of endogenous dopamine by stimulation of 5-hydroxytryptamine3 receptors in rat striatum. J Pharmacol Exp Ther 1989; 251: 803–9.
Porras G, De Deurwaerdère, Moison D, Spampinato U . Conditional involvement of striatal serotonin3 receptors in the control of in vivo dopamine outflow in the rat striatum. Eur J Neurosci 2003; 17: 771–81.
Wu J, Liu Q, Yu K, Hu J, Kuo YP, Segerberg M, et al. Roles of nicotinic acetylcholine receptor β subunits in function of human α4-containing nicotinic receptors. J Physiol 2006; 576: 103–18.
Samanin R, Quattrone A, Peri G, Ladinsky H, Consolo S . Evidence of an interaction between serotoninergic and cholinergic neurons in the corpus striatum and hippocampous of the rat brain. Brain Res 1978; 151: 73–83.
Acknowledgements
This work was supported by a grant from the NIH (No AG21586) and the State of Pennsylvania Tobacco Formula Funds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dougherty, J., Nichols, R. Cross-regulation between colocalized nicotinic acetylcholine and 5-HT3 serotonin receptors on presynaptic nerve terminals. Acta Pharmacol Sin 30, 788–794 (2009). https://doi.org/10.1038/aps.2009.62
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2009.62
Keywords
This article is cited by
-
Cholinergic regulation of mood: from basic and clinical studies to emerging therapeutics
Molecular Psychiatry (2019)
-
Additive interaction between scopolamine and nitric oxide agents on immobility in the forced swim test but not exploratory activity in the hole-board
Psychopharmacology (2019)
-
How Serotonin is Related with Lower Urinary Dysfunction
Advances in Therapy (2015)
-
Enhanced alcohol-seeking behavior by nicotine in the posterior ventral tegmental area of female alcohol-preferring (P) rats: modulation by serotonin-3 and nicotinic cholinergic receptors
Psychopharmacology (2014)
-
Association and interaction analyses of 5-HT3 receptor and serotonin transporter genes with alcohol, cocaine, and nicotine dependence using the SAGE data
Human Genetics (2014)
