
Abstract
Familial adenomatous polyposis coli (FAP) has been shown to be associated with germline mutations of the adenomatous polyposis gene (APC) on chromosome 5. Extra-colonic manifestations also occur in FAP and include desmoid tumors, epidermoid cysts and osteomas. The combination of FAP with extracolonic symptoms is commonly referred to as Gardner’s sydrome. It remains difficult, however, to predict which patients may have a propensity to develop extracolonic manifestations. The rapid acetylation phenotype is believed to be associated with an increased likelihood of sporadic colorectal cancer, whereas the slow acetylation phenotype is recognized as a predisposing factor for bladder cancer. The slow acetylation phenotype is caused by mutant alleles of the cytosolic enzyme N-acetyltransferase (NAT2). In this study, we determined the NAT2 genotype in members of one large FAP family and three smaller ones all of which had been shown to harbor the same germline APC gene mutation. We observed a significant correlation between slow acetylation genotypes and extracolonic manifestations of the disease. Rapid acetylation genotypes were not overrepresented in colorectal cancer cases in this family as compared to the frequency of this genotype in the normal Caucasian population.
Similar content being viewed by others
Introduction
Familial adenomatous polyposis (FAP) is an autosomal-dominantly inherited disease affecting approximately 1 in 10,000 people [1] and is characterized by the development of hundreds to thousands of colorectal polyps [2], which if left untreated would almost certainly develop into colorectal cancer [3]. In association with FAP there exists a more severe form of the disease, otherwise known as Gardner’s syndrome, which includes extracolonic manifestations such as osteomas, epidermoid cysts, small intestinal carcinomas, gastric polyps, thyroid carcinomas and dental abnormalities [4]. However, a precise definition of this entity is lacking. Common to both forms of the disease is the increased likelihood to develop desmoid tumors which appear to be a significant side effect after surgery [5].
The FAP locus was identified after the observation of a constitutional interstitial deletion on chromosome 5q in a mentally retarded patient with FAP and a desmoid tumor [6] followed by linkage analysis to the 5q21–22 locus [7]. More recently, the gene involved in the inheritance of FAP has been sequenced and is known as the APC gene [8, 9]. Many APC germline mutations have been found in FAP patients and in presymptomatic carriers [10–15]. Of particular note is that identical APC gene mutations have been identified in unrelated and related FAP families presenting with or without extracolonic manifestations [16, 17]. The most likely explanation is the presence of a modifying gene that is able to influence disease expression. Support for the existence of a modifying locus comes from studies using the naturally occurring multiple intestinal neoplasia (Min) mouse model [18]. More recently, a candidate modifier gene has been identified in the mouse, namely secretory phospholipase A2 [19]. This gene does not, however, appear to be associated with phenotypic modification in man as in both FAP families and sporadic colorectal cancer patients no association has been identified [20, 21].
It has been recognized that acetylation catalyzed by the cytosolic enzyme N-acetyltransferase (NAT) is an important process in the activation of arylamine carcinogens and that there exists genetic variation within the population, such that there are rapid and slow acetylators [22]. The two polymorphic isozymes NAT1 and NAT2 perform the function of arylamine N-acetylation and O-acetylation of hydroxyamines to yield acetoxy derivatives [23, 24], respectively. It has been shown that only the isoenzyme NAT2 is responsible for the acetylation polymorphism [25]. Six point mutations of the NAT2 gene on chromosome 8 have been identified which either occur as single mutations or in combination. They produce over 10 mutant alleles of the NAT2 gene. Three alleles (M1, M2, M3) account for over 95% of the slow acetylation phenotype in most populations [26–29].
The frequencies of rapid and slow acetylators vary remarkably in different ethnic populations. In Europe and North America there are about 40–70% slow acetylators, compared to only 10–20% in Japan and China [30].
Two studies have shown that sporadic colorectal cancer (CRC) tends to be associated with a rapid acetylator phenotype [31–33] which is believed to be a predisposing factor in the development of the disease. The slow acetylator phenotype is overrepresented in patients with bladder cancer that were exposed to amine carcinogens [34–36].
In this study we examined one large kindred with FAP which included family members with extracolonic manifestations and three smaller families. Previously, we have shown that all affected members of the large kindred have inherited the same germline mutation in the APC gene [17]. Three additional smaller families not known to be related to the large family at the time of the analysis were also included in the study as they too harbored the same APC germline mutation. Subsequent haplotype and pedigree analysis of all four families has since revealed that they are related sharing a common ancestor in the early 18th century. Genotyping analysis of the NAT2 gene revealed that persons classified for this study as suffering from either extra-colonic disease alone or in combination with colonic disease, appeared to correlate with a particular acetylation status. In contrast, no difference between the frequency of slow or rapid acetylators in the subgroup suffering from colonic disease alone and the general population was observed.
Materials and Methods
Patients
Symptomatic patients were first ascertained at clinical presentation, thereafter relatives were contacted through their family doctor after the index patients had filled out a questionnaire pertaining to FAP in their family. The clinical diagnosis of FAP was based on colonoscopy. When only colonic symptoms were evident they were designated as FAP patients, whereas those patients who had any other signs before or after colonic disease were referred to for this analysis as having extracolonic disease plus or minus FAP.
DNA Isolation
Genomic DNA was isolated from EDTA blood according to the method described by Miller et al. [37].
Detection of the NAT2 Genotype by PCR Amplification
The specific amplification of the NAT2 alleles was carried out in separate PCR reactions for the wild-type and mutant alleles of M1, M2 and M3. The primers and amplification procedures used have been described previously [26]. Persons that were homozygous or heterozygous for a wild-type allele were designated as having a rapid acetylation genotype. Those being homozygous for one or compound heterozygous for two mutated NAT2 alleles were designated as slow acetylators. In a single case the genotype of one individual was deduced by pedigree analysis of the first-degree relatives and that of the spouse. Precise studies have shown that the phenotype can be predicted by these techniques in over 90% of the cases [38].
Statistical Analysis
Fisher’s exact test and a binomial test were used to determine differences between FAP patients and patients with extracolonic disease ± FAP.
Results
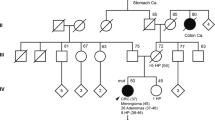
A detailed pedigree of the very large family used for this investigation has been reported previously [17]. A condensed version of the family is shown in figure 1 which indicates only the most important branches of the family, age of diagnosis and acetylation genotype. Every affected person in all four families harbored the same germline APC mutation. Table 1 provides a brief profile of disease phenotypes and NAT2 genotypes. Asymptomatic APC gene mutation carriers were excluded from any calculation as it is impossible to predict what disease phenotype they will eventually express.

Abridged version of the large kindred indicating age of onset and genotype analysis in persons shown. Checkered symbols denote EC ± FAP phenotype and black symbols denote FAP.
From a total of 127 persons, 52% were rapid acetylators and 48% were slow acetylators.
A comparison of NAT2 genotypes between all affected APC gene carriers and healthy controls revealed no deviation from independence (table 2) implying that apparent differences between the frequencies of the slow/rapid genotypes within the two groups were likely due to chance alone. If the gene carrier group was further subdivided into patients with FAP alone and patients with extracolonic disease ± FAP, several differences are observed. Between the control group and FAP no significant difference was seen (p = 0.172). When the extracolonic disease group was compared to the control group a significant difference between the presence of slow and fast acetylators was observed (p = 0.022, data shown in table 3). Finally, comparing the extracolonic disease group to the FAP group, a highly significant difference was observed (p = 0.004) between the two groups in that slow acetylation appeared to be almost exclusively associated with extracolonic disease. In summary, the extracolonic disease group has a markedly different acetylation genotype than either the FAP or healthy controls.
One problem with the above analysis is that it assumes that the genotype of the patients under investigation are independent of one another. Since it is not clear how deviations from independence will affect the analysis or interpretation of the results, other tests that take this into account need to be employed. In an attempt to overcome these shortcomings, we have compared the NAT2 genotypes of patients who are the offspring of matings between compound heterozygotes or homozygotes for M1, M2, M3 and heterozygotes for W (table 4) and therefore have a 50% probability of inheriting the W allele. Although the numbers are small, it is noticeable that all the extracolonic disease patients were slow acetylators (p = 0.03 for a binomial test of deviation from 50:50 expectation), whereas the FAP patients conformed to expected values from such crosses.
Together, the results imply that there is a relationship between slow acetylation and the likelihood of extracolonic disease development due to the paucity of rapid acetylators in the extracolonic disease subgroup.
Discussion
Of considerable interest to the clinician would be the possibility of identifying an additional genetic or environmental factor that may predispose a person already harboring an APC mutation to the development of extracolonic disease [39, 40]. In this report we have attempted to correlate the acetylation genotype to the likelihood of extracolonic disease development.
The existence of families that carry the same germline mutation in the APC gene yet present with markedly different phenotypes is indicative of other influences affecting disease expression. Strong candidate genes that may modify disease expression include enzymes that are associated with carcinogen metabolism, such as NAT2. In this report four families are presented where affected persons all carry the same germline mutation, yet present with markedly varied disease phenotypes. Therefore, differences in disease expression cannot be attributed to mutations occurring at different sites within the APC gene which has previously been shown in some cases to be correlated with disease presentation. Together this implies that the most likely cause of disease diversity is the influence of one or more modifying genes. One modifier gene, phospholipase A2, has recently been described which appears in mice to alter the number of polyps and age of onset of colonic disease [19]. Recent evidence indicates, however, that this gene is not associated with phenotypic variation in man [20, 21]. It cannot be excluded, however, that the diversity of phenotype seen in families harboring truncating 3′ APC germline mutations is due to the instability of the resulting prematurely terminated protein product which results in a null APC allele. The presence of a null APC allele implies that APC carcinogenesis is driven via haplo insufficiency and is not due to the effects of a protein exhibiting only partial function [41]. This effect may not be seen in families where the more common APC mutations have been identified as these would lead to a putative dominant negative effect and hence could negate any influence brought about by a modifying factor. Thus it is possible that the NAT2 slow acetylator genotype represents a modifier of null APC mutations and not necessarily of the more common APC mutations that lead to a stable truncated protein.
In an attempt to determine if other factors were involved in the expression of extracolonic manifestations of FAP, a difference in the acetylation capacity of persons carrying a specific germline mutation in the APC gene was considered. Previously, it has been shown that there is an apparent association between rapid acetylation and sporadic colorectal cancer [31–33]. Conversely, the slow acetylator phenotype is overrepresented in patients with bladder cancer [34–36], indicating that the two different environments may be important with respect to disease expression. Bladder cancer has not been reported in any of the patients used in this investigation.
In the present study, we subdivided all clinically verified patients with an APC gene mutation into either colorectal cancer alone (FAP) or colorectal cancer in association with extracolonic manifestations that occurred before or after colonic disease. The original description of FAP includes desmoid tumors. Desmoid tumors are relatively common complications in FAP patients after colonic surgery and are often associated with FAP and are not necessarily present in persons who exhibit other extracolonic symptoms [5]. For the purposes of the current study patients who had developed desmoid tumors after surgery were classified as having extracolonic disease as we wanted to determine if patients who developed any extracolonic lesions before or after colorectal disease carried an additional predisposition. Interestingly, most patients within the extracolonic disease group presented initially with an extracolonic manifestation (such as osteomas or epidermal cysts) at or before colonoscopy.
From this family study, we found that the slow acetylation phenotype appears to be associated with the development of extracolonic disease in persons predisposed to the development of polyposis coli. The lack of association between rapid acetylators and persons expressing only FAP is not surprising. The most likely explanation for this observation is that persons who carry a germline mutation of the APC gene are at such an increased risk of developing colorectal cancer that acetylation is unlikely to play a significant role in CRC development.
The question arises as to how acetylation status plays a role in the expression of extracolonic disease. It has been documented that there is an association of rapid acetylation and colorectal cancer [31–33] and that the predisposition towards colorectal cancer is due to the rapid acetylation of aryl amines in the liver and gut such that few if any of these carcinogenic compounds reach other organs. An intriguing aspect is the association of slow acetylation and bladder cancer presumably due to a higher concentration of aryl amines in the bladder which could act on the bladder mucosa. Similarly, it could be expected that in slow acetylators, aryl amines may be present at a higher concentration within the body such that they may affect sensitive tissues and predispose an APC gene carrier to extracolonic disease. As the colon has been shown to be a major site of acetylation in comparison with the liver [30], it is possible that after colectomy there is a reduction in acetylation capacity. Alternatively, heterogeneity of the acetylation phenotype may exist within each individual thus leading to different clinical outcomes. One aspect that could not be addressed in this study concerns those slow acetylators who had thus far only presented with colorectal disease. Will they develop other symptoms associated with polyposis coli? Furthermore, it is possible that the rapid acetylators develop extracolonic disease as they get older and thus eliminate any differences between rapid and slow acetylators with respect to extracolonic disease development. The evaluation of age adjusted phenotype classes would better define the role of NAT2 in extracolonic disease development. These questions can only be answered by continued surveillance and more rigorous screening for other manifestations of the disease.
In conclusion, we have demonstrated in one large family and several smaller ones all carrying the same APC germline mutation an association of slow acetylation genotype and extracolonic disease. Due to the paucity of cases it is not possible to proceed much further with this analysis in our study population. Furthermore, it cannot be ruled out that the association described here is peculiar to the APC gene mutation found in these families. Therefore, to confirm that slow acetylators may be associated with an increased risk of extracolonic disease, additional studies are required using as many clinically and genetically verified cases as possible. This we believe could only be performed with a large multicenter study.
References
Reed TE, Neel JV: A genetic study of multiple polyposis of the colon (with appendix deriving a method of estimating relative fitness). Am J Hum Genet 1955;7:236–263.
Bülow S: Familial polyposis coli. Dan Med Bull 1987;34:1–5.
Bussey HJR: Familial Polyposis coli: Family Studies, Histopathology, Differential Diagnosis and Results of Treatment. Baltimore, Johns Hopkins University Press, 1975.
Gardner EJ: A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum. Am J Hum Genet 1951;3:167–176.
Klemmer S, Passkey L, Daces J: Occurrence of desmoids in patients with familial adenomatous polyposis of the colon. Am J Med Genet 1987;28:385–392.
Herrera L, Kakati S, Gibas L, Pietrzak E, Sandberg AA: Gardner syndrome in a man with an interstitial deletion of 5q. Am J Med Genet 1986;25:473–476.
Bodmer WF, Bailey CJ, Bodmer J, Bussey HJR, Ellis A, Gorman P, Lucibello FC, Murday VA, Rider SH, Scambler P, Sheer D, Solomon E, Spurr NK: Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 1987;328:614–616.
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, Sargeant L, Krapcho K, Wolff E, Burt R, Hughes JP, Warrington J, McPherson J, Wasmuth J, Le Paslier D, Abderrahin H, Cohen D, Leppert M, White R: Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991; 66: 589–600.
Kinzler KW, Nilbert MC, Su L-K, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, Finniear R, Markham A, Groffen J, Boguski MS, Altschul SF, Haar A, Ado H, Miyoshi Y, Midi Y, Nishisho I, Nakamura Y: Identification of FAP locus genes from chromosome 5q21. Science 1991; 253:661–665.
Fodde R, van der Luijt R, Wijnen J, Tops C, van der Klift H, van Leeuwen-Cornelisse I, Griffioen G, Vasen H, Merra Khan P: Eight novel inactivating germline mutations at the APC gene identified by denaturing gradient gel electrophoresis. Genomics 1992; 13:1162–1168.
Miyoshi Y, Ado H, Nagase H, Nishisho I, Haar A, Midi Y, Mori T, Utsunomiya J, Baba S, Petersen G, Hamilton SR, Kinzler KW, Vogelstein B, Nakamura Y: Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci USA 1992; 89:4452–4456.
Groden J, Gelbert L, Thliveris A, Nelson L, Robertson M, Joslyn G, Samowitz W, Spirio L, Carlson M, Burt R, Leppert M, White R: Mutational analysis of patients with adenomatous polyposis: Identical inactivating mutations in unrelated individuals. Am J Hum Genet 1993; 52:263–272.
Varesco L, Gismondi V, James RM, Robertson M, Grammatico P, Groden J, Casarino L, De Benedetti L, Bafico A, Bertareio L, Sala P, Sassatelli R, Ponz de Leon M, Biasco G, Allegretti A, Aste H, De Sanctis S, Rossetti C, Illeni MT, Sciarra A, Del Porto G, White R, Ferrara GB: Identification of APC gene mutations in Italian adenomatous polyposis coli patients by PCR-SSCP analysis. Am J Hum Genet 1993;52: 280–285.
Olschwang S, Laurent-Puig P, Groden J, White R, Thomas G: Germ-line mutations in the first 14 exons of the adenomatous polyposis coli (APC) gene. Am J Hum Genet 1993;52:273–279.
Dobbie Z, Spycher M, Mary J-I, Haner J, Guldenschuh I, Hürlimann R, Ammann R, Roth J, Müller HJ, Scott RJ: Correlation between the development of extracolonic manifestations in FAP patients and mutations beyond codon 1403 in the APC gene. J Med Genet 1996;33: 274–280.
Nishisho I, Nakamura Y, Miyoshi Y, Midi Y, Ado H, Haar A, Koyama K, Utsunomiya J, Baba S, Hedge P, Markham A, Krush AJ, Petersen G, Hamilton SR, Nilbert MC, Levy DB, Bryan TM, Preisinger AC, Smith KJ, Su LK, Kinzler KW, Vogelstein B: Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991;253:665–669.
Scott RJ, van der Luijt R, Spycher M, Mary J-L, Müller A, Hoppeler T, Haner M, Müller HJ, Martinoli S, Brazzola P-L, Meera Khan P: Novel germline mutation in a large familial adenomatous polyposis kindred displaying variable phenotypes. Gut 1995;36:731–736.
Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W: Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell 1994;75:631–639.
Macphee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM: The secretory phospholipase A2 gene is a candidiate for the Mom1 locus, a major modifier of Apcmin-induced intestinal neoplasia. Cell 1995;81: 957–966.
Dobbie Z, Müller HJ, Scott RJ: Secretory phospholipase A2 does not appear to be associated with phenotypic variation in familial adenomatous polyposis. Hum Genet 1996;98:396–390.
Riggins GJ, Markowitz S, Wilson JK, Vogelstein B, Kinzler KW: Absence of secretory phospholipase A2 gene alterations in human colorectal cancer. Cancer Res 1995;55:5184–5186.
Weber WW, Hein DW: Acetylator genotype and arylamine carcinogenesis. Biochim Biophys Acta 1985;948:37–66.
Bock KW: Metabolic polymorphisms affecting activation of toxic and mutagenic arylamines. TIPS 1992;13:223–226.
Bell DA, Badawar AF, Lang NP, Ilett KF, Hirvonen A: Polymorphism in the N-acetyltransferase 1 (NAT1) polyadenylation signal: Association of NAT1*10 allele with higher N-acetylation activity in bladder and colon tissue. Cancer Res 1995;55:5226–5229.
Grant D, Blum M, Beer M, Meyer U: Monomorphic and polymorphic human arylamine N-acetyltransferase: A comparison of isozymes and expressed products of two cloned genes. Mol Pharm 1990;39:184–191.
Blum M, Demierre A, Grant DM, Heim M, Meier UA: Molecular mechanism of slow acetylation of drugs and carcinogens in humans. Proc Natl Acad Sci USA 1991;88:5237–5241.
Vatsis KP, Martell KJ, Weber W: Diverse point mutations in the human gene for polymorphic N-acetyltransferase. Proc Natl Acad Sci USA 1991;88:6333–6337.
Hickman D, Sim E: N-acetyltransferase polymorphism: Comparison of phenotype and genotype in humans. Biochem Pharmacol 1991; 42:1007–1014.
Lin HJ, Han C-Y, Lin BK, Hardy S: Slow acetylator mutations in the human polymorphic N-acetyltransferase gene in 786 Asians, blacks, hispanics, and whites: Application to metabolic epidemiology. Am J Hum Genet 1993;52:827–834.
Evans DAP: N-acetyltransferase. Pharmacol Ther 1989;42:157–234.
Wohleib JC, Hunter CE, Blass B, Kadlubar FF, Chu DZJ, Lang NP: Aromatic amine acetyltransferase as a marker for colorectal cancer: Environmental and demographic associations. Int J Cancer 1990;46:22–30.
Ilett KF, David BM, Detchon P, Castleden WM, Kwa R: Acetylation phenotype in colorectal cancer. Cancer Res 1987;47:1466–1469.
Lang NP, Butler MA, Massengill J, Lawson M, Stotts RC, Hauer-Jensen M, Kadlubar FF: Rapid metabolic phenotypes for acetyltransferase and cytochrome P4501A2 and putative exposure to food-borne heterocyclic amines increase the risk for colorectal cancer of polyps. Cancer Epidemiol Biomarkers Prevent 1994;3: 675–682.
Cartwright RA, Glashan RW, Rogers HJ, Ahmed RA, Hall DB, Higgins E, Khan MA: The role of N-acetyltransferase phenotypes in bladder cancer: A pharmacogenetic epidemiological approach to bladder cancer. Lancet 1982;ii:842–846.
Wolf H, Lower GM Jr, Bryan GT: Role of N-acetyltransferase phenotype in human susceptibility to bladder carcinogenic arylamines. Scand J Urol Nephrol 1980; 14:161–165.
Evans DAP, Eze LC, Whibley EJ: The association of the slow acetylator phenotype with bladder cancer. J Med Genet 1983;20:330–333.
Miller SA, Dykes DD, Polesky HF: A salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res 1988; 16:739.
Graf T, Broly F, Hoffman F, Probst M, Meyer UA, Howald H: Prediction of phenotype for acetylation and for debrisoquine hydroxylation by DNA tests in healthy human volunteers. Eur J Clin Pharmacokinet 1992;43:399–403.
Scott RJ, Müller HJ: Familial and genetic aspects of colorectal carcinogenesis. Eur J Cancer 1993;29A:2163–2167.
Müller HJ, Scott RJ, Weber W, Meier R: Colorectal cancer: Lessons for genetic counselling and care for families. Clin Genet 1994;46:106–114.
van der Luijt RB, Meera Khan P, Vasen HFA, Breukel C, Tops CMJ, Scott RJ, Fodde R: Germline mutations in the 3′ part of APC exon 15 do not result in truncated proteins and are associated with attenuated adenomatous polyposis coli. Hum Genet 1996;98:727–734.
Acknowledgements
This work is supported in part by grants from the Swiss National Foundation No. 3200-042558.94 and the Swiss Cancer League AKT 78.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Scott, R.J., Taeschner, W., Heinimann, K. et al. Association of Extracolonic Manifestations of Familial Adenomatous Polyposis with Acetylation Phenotype in a Large FAP Kindred. Eur J Hum Genet 5, 43–49 (1997). https://doi.org/10.1007/BF03405876
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF03405876
