
Abstract
The cancer-predisposing syndrome caused by biallelic mutations in NTHL1 may not be a solely colorectal cancer (CRC) and polyposis syndrome but rather a multi-tumor recessive disease. The presence of ≤10 adenomas in several mutation carriers suggests a possible causal role of NTHL1 in hereditary or early-onset nonpolyposis CRC. The involvement of NTHL1 in serrated/hyperplastic polyposis remains unexplored. The aim of our study is to elucidate the role of NTHL1 in the predisposition to personal or familial history of multiple tumor types, familial/early-onset nonpolyposis CRC, and serrated polyposis. NTHL1 mutational screening was performed in 312 cancer patients with personal or family history of multiple tumor types, 488 with hereditary nonpolyposis CRC, and 96 with serrated/hyperplastic polyposis. While no biallelic mutation carriers were identified in patients with personal and/or family history of multiple tumor types or with serrated polyposis, one was identified among the 488 nonpolyposis CRC patients. The carrier of c.268C>T (p.Q90*) and 550-1G>A was diagnosed with CRC and meningioma at ages 37 and 45 respectively, being reclassified as attenuated adenomatous polyposis after the cumulative detection of 26 adenomas. Our findings suggest that biallelic mutations in NTHL1 rarely cause CRC, a personal/familial multi-tumor history, or serrated polyposis, in absence of adenomas.
Similar content being viewed by others
Introduction
In 2015 Weren et al. described a hereditary cancer syndrome caused by biallelic mutations in the DNA base excision repair gene NTHL1, characterized by attenuated adenomatous polyposis and increased colorectal cancer (CRC) risk, largely resembling the recessive syndrome caused by MUTYH mutations1. To date, 33 homozygous or compound heterozygous NTHL1 mutation carriers have been reported (21 families)1,2,3,4,5,6,7,8. More than 5 colonic adenomas (range: 6 to >50) were identified in 24 of the 28 (85%) mutation carriers who underwent colonoscopy screening, and CRC was diagnosed in 19 (68%) of them. Noteworthy, 17 carriers (57%) were diagnosed with multiple primary malignant tumors in extracolonic locations, being the most recurrently found breast and endometrial tumors, head neck squamous cell carcimomas, meningiomas, and bladder and basal cell carcinomas, suggesting that the NTHL1-associated syndrome is a multi-tumor disease rather than a solely CRC syndrome. On the other hand, the fact that at least ¼ (7/28) of the reported biallelic mutation carriers who underwent colonoscopy screening had ≤10 adenomas, and that ≥5 hyperplastic polyps were detected in five carriers (polyp number range: 5–>30), lead us to suspect a possible association of NTHL1 mutations with nonpolyposis CRC and serrated/hyperplastic polyposis.
Based on previous evidence and with the aim of refining the phenotypic characteristics of the NTHL1-associated syndrome, here we evaluated the implication of NTHL1 biallelic mutations in the predisposition to personal or familial history of multiple tumor types, familial/early-onset nonpolyposis CRC, and serrated/hyperplastic polyposis.
Materials and Methods
Patients
In order to evaluate the role of NTHL1 biallelic mutations in the context of a multi-tumor syndrome, we studied 312 unrelated cancer-affected patients who fulfilled the selection criteria indicated in Supplementary Table 1 and without mutations in the high penetrance genes associated with the corresponding phenotypes. Briefly, we included cancer-affected individuals with personal or familial history of: (i) colorectal, endometrial, small intestine or gastric cancer AND breast or ovarian cancer (n = 122); (ii) brain cancer AND any other tumor (n = 22); (iii) breast, endometrial, brain or skin cancer AND > 5 adenomatous or hyperplastic polyps (classic and attenuated familial adenomatous polyposis excluded) (n = 34); (iv) multiple primary tumors (excluding the combinations indicated in the previous categories) (n = 30); and (v) patients fulfilling the clinical criteria for germline TP53 mutational screening but with no mutations in the gene (Li-Fraumeni) (n = 104).
A total of 488 hereditary nonpolyposis CRC cases (473 families) were also included in the study5. Nonpolyposis cases were mismatch repair (MMR)-proficient, i.e., their tumors showed microsatellite stability and expression of the MMR proteins MLH1, MSH2, MSH6, and PMS2. All tested individuals were affected with cancer, 96.3% with CRC. The mean age at cancer diagnosis was 49 (range: 16–82). Among the 473 studied families, 58 (12.2%) fulfilled the Amsterdam criteria, 385 (81.4%) the Bethesda guidelines, and 30 (6.3%), none the established criteria for hereditary nonpolyposis CRC. Detailed description of the hereditary nonpolyposis CRC cases is shown in Supplementary Table 2.
Also, 96 individuals diagnosed with hyperplastic/serrated polyposis9, were screened for germline mutations in NTHL1. Description of this cohort is shown in Supplementary Table 3.
All patients were assessed at the Hereditary Cancer Program of the Catalan Institute of Oncology (Spain) between 1999 and 2017. Informed consent was obtained from the participants and all methods were performed in accordance with relevant guidelines and regulations. The study received the approval of the Ethics Committee of the Institut d’Investigació Biomèdica de Bellvitge (IDIBELL).
Mutational screening of NTHL1
NTHL1 c.268C > T (p.Q90*) was genotyped in all patients by using a KASP genotyping assay (LGC Genomics, Hoddesdon, UK) in a LightCycler® 480 system (Roche, Basel, Switzerland).
Mutational screening of protein-coding exons and flanking sequences (+/−20 base pairs) was performed by direct automated (Sanger) sequencing in the 312 cancer patients with familial/personal history of multiple tumor types and in the 96 individuals with hyperplastic/serrated polyposis. Sequencing was performed at STAB VIDA (Caparica, Portugal) and data was analyzed with SeqMan Pro (Lasergene 13, DNASTAR, Madison, WI) and/or Mutation Surveyor v.3.10 (SoftGenetics, State College, PA). The primers used for amplification and sequencing are shown in Supplementary Table 4.
Hereditary nonpolyposis CRC patients were screened for NTHL1 mutations using a combination of PCR amplification in pooled DNAs and targeted massively parallel sequencing, as previously described10, using the amplification primers listed in Supplementary Table 4. Sanger sequencing of the affected exon was performed to identify the mutated individuals among the samples included in the corresponding DNA pool.
In silico predictions
The impact of missense variants at the protein level was analyzed using the in silico algorithms PolyPhen-2, SIFT, CONDEL, Mutation Taster and Align GVGD11,12,13,14,15. The potential effects on splicing were evaluated by using Human Splice Finder v.3.016. Except for CONDEL, prediction data were provided by Alamut Visual v2.7.1 (Interactive Biosoftware, Rouen, France).
PCR amplicon cloning
Exons 2–4 (2,985 bp) were PCR amplified in the gDNA extracted from the carrier of NTHL1 c.268C>T and c.550-1G>A, cloned into the pGEM-T Easy Vector System (Promega, Madison, Wisconsin, USA) and transformed into JM109 competent cells (Promega). Individual colonies were isolated, PCR amplified and sequenced.
NTHL1 constitutional methylation
Methylation of the promoter of NTHL1 was analyzed by means of methylation-specific melting curve analysis (MS-MCA) on bisulfite-modified DNA (EZ DNA Methylation-Gold Kit, Zymo Research, Orange, CA, USA). Thirty-eight and 17 CpG sites located in the NTHL1 promoter region were evaluated in two independent assays. Methylation status was determined comparing melting curves obtained from samples versus controls, which included the CpG methylated Jurkat genomic DNA as methylated control, and DNA amplified by whole genome amplification as non-methylated control. Primer sequences are shown in Supplementary Table 4.
NTHL1 copy number alterations
Assessment of copy number variation in the NTHL1 region was carried out using the Illumina Infinium Global Screening Array v2.0. Data on hybridization intensity for each probe were used to calculate the Log R ratio (LRR) and B allele frequency (BAF) values. These values were used to identify copy number regions and to segment the genome. Segmentation calculations were performed with genoCN software17 using the R package.
Results and Discussion
No biallelic NTHL1 mutation carriers were identified among the 312 patients with familial/personal history of multiple tumor types (polyposis and patients fulfilling the clinical criteria for hereditary nonpolyposis CRC cancer, were excluded). The c.268C>T (p.Q90*) recurrent pathogenic mutation, c.444G>A (p.A148=) and c.527 T>C (p.I176T), were identified in heterozygosis in 3 unrelated patients (Supplementary Table 5).
NTHL1 mutational screening in the 488 MMR-proficient hereditary colorectal cancer patients (473 families), which included 58 families fulfilling the Amsterdam criteria, identified one compound heterozygous of c.268C>T (p.Q90*) and c.550-1G>A (Table 1). No additional relatives were available for segregation analysis. Cloning of the PCR product that included the two variants confirmed the biallelic nature of the mutations; i.e., they were found to be in different alleles, in trans (Supplementary Fig. 1). In addition to the biallelic mutation carrier, one heterozygous carrier of c.268C>T (p.Q90*), one of c.550-1G>A, one of c.793G>A (p.A265T) and two of c.527T>C (p.I176T), were identified (Supplementary Table 6). NTHL1 was re-sequenced in the five heterozygotes in order to assess the presence of a second mutation, but none was detected. The two carriers of c.268C>T (p.Q90*) in the MMR-proficient nonpolyposis CRC series had been previously identified by our group by mutation-specific genotyping5. However, in that study, sequencing of the complete coding region of NTHL1 in the two carriers failed to identify in one of them the herein reported second NTHL1 mutation, c.550-1G>A, possibly due to allele dropout.
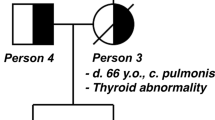
The biallelic carrier of the NTHL1 pathogenic mutations c.[268C>T]; [550-1G>A] was diagnosed with CRC and 3 adenomas at age 37, thus fulfilling the Bethesda criteria for hereditary CRC. Review of her updated clinical history revealed that annual/biennial colonoscopy screenings between ages 37–46 (8 colonoscopies) detected a total of 26 adenomas and 8 hyperplastic polyps, which now leads to a reclassification as attenuated polyposis. Moreover, she was diagnosed with a meningioma at 45 years of age (Family pedigree in Fig. 1).

Pedigree of the carrier of NTHL1 c.[268C>T];[550-1G>A]. Filled black symbol, cancer affected. The black arrow is pointing out the index case. Ages at information gathering or at death, when available, are indicated on the top-right corner, and ages at cancer diagnosis, after tumour type. Abbreviations: ca, cancer; CRC, colorectal cancer; HP, hyperplastic polyps; mut, NTHL1 biallelic carrier.
Broderick et al. analyzed NTHL1 from whole-exome sequencing data performed in 863 unexplained familial/early-onset CRC cases, finding no biallelic mutations in nonpolyposis CRC patients (biallelic NTHL1 mutations were identified in a 41 year-old patient diagnosed with CRC and polyposis)3. Likewise, Zhang et al. sequenced the NTHL1 gene in 140 patients diagnosed with CRC before 35 years of age (from an unselected series of >2,300 Chinese CRC patients), finding no pathogenic mutations in the gene18. The additive evidence gathered in these two recent studies and ours (total number of patients studied: 1,491; number of NTHL1 biallelic carriers: 1, who has been reclassified as having attenuated adenomatous polyposis) suggests that the contribution of biallelic NTHL1 mutations to nonpolyposis CRC is null or, at most, negligible.
On the other hand, no NTHL1 mutations were identified among the 96 serrated polyposis patients, either in homozygosis or heterozygosity. Although our findings need to be validated in larger cohorts, they suggest that biallelic NTHL1 mutations are not a major cause of serrated polyposis, in spite of the detection of several hyperplastic polyps (range: 5->30) in 6 out of 34 NTHL1 biallelic mutation carriers reported to date, including the c.[268C>T]; [c.550-1G>A] carrier identified in the current study, with 26 adenomas and 8 hyperplastic polyps detected before age 50.
In the herein studied cohorts, the frequency of heterozygous missense, splice-site and loss-of-function variants (population minor allele frequency (MAF) < 1%) in NTHL1 (NM_002528.5): 3/312 (0.96%) in patients with personal and/or familial history of multiple tumor types, 5/487 (1.03%) (5/472 (1.06%) if only unrelated patients are considered) in hereditary nonpolyposis CRC - the NTHL1-mutated attenuated adenomatous polyposis patient excluded-, and 0/96 (0%) in hyperplastic/serrated polyposis, is not higher than the frequency identified in European cancer-free population, i.e. 1.74% (75/4299 individuals; source: NHLBI GO Exome Sequencing Project http://evs.gs.washington.edu/EVS/), a priori supporting the lack of association of monoallelic NTHL1 mutations with cancer predisposition.
Although never reported before, we assessed the presence of NTHL1 promoter hypermethylation in the identified monoallelic mutation carriers in order to discard the presence of other types of alterations affecting the -a priori- wildtype allele. None of the eight heterozygotes showed constitutional NTHL1 CpG island methylation (Supplementary Fig. 2). Copy number alteration analysis could be performed in six of the eight monoallelic mutation carriers identified, finding no large deletions or rearrangements affecting the gene. Of note, neither our study nor previous analyses reported so far have included the study of large deletions or constitutional epimutations in NTHL1 in the complete series, which might have caused the under-detection of mutations in the studied cohorts.
In conclusion, our findings suggest that: (i) the NTHL1-associated syndrome is not a multi-tumor syndrome in absence of adenomatous polyposis, (ii) the serrated polyposis syndrome is not caused by NTHL1 mutations, and (iii) the presence of biallelic NTHL1 mutation carriers in hereditary CRC patients without polyposis is, if any, extremely rare.
References
Weren, R. D. et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 47, 668–671 (2015).
Rivera, B., Castellsague, E., Bah, I., van Kempen, L. C. & Foulkes, W. D. Biallelic NTHL1 Mutations in a Woman with Multiple Primary Tumors. N Engl J Med 373, 1985–1986 (2015).
Broderick, P. et al. Validation of Recently Proposed Colorectal Cancer Susceptibility Gene Variants in an Analysis of Families and Patients-a Systematic Review. Gastroenterology 152, 75–77 e4 (2017).
Chubb, D. et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nature communications 7, 11883 (2016).
Belhadj, S. et al. Delineating the Phenotypic Spectrum of the NTHL1-Associated Polyposis. Clin Gastroenterol Hepatol 15, 461–462 (2017).
Fostira, F. et al. Extending the clinical phenotype associated with biallelic NTHL1 germline mutations. Clinical genetics 94, 588–589 (2018).
Grolleman, J. E. et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-tumor Phenotype. Cancer cell 35, 256–266 e255 (2019).
Groves, A., Gleeson, M. & Spigelman, A. D. NTHL1-associate polyposis: first Australian case report. Familial cancer 18, 179–182 (2019).
Quintana, I. et al. Evidence suggests that germline RNF43 mutations are a rare cause of serrated polyposis. Gut 67, 2230–2232 (2018).
Puente, X. S. et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 475, 101–105 (2011).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nature methods 7, 248–249 (2010).
Gonzalez-Perez, A. & Lopez-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. American journal of human genetics 88, 440–449 (2011).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols 4, 1073–1081 (2009).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nature methods 11, 361–362 (2014).
Mathe, E. et al. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic acids research 34, 1317–1325 (2006).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids research 37, e67 (2009).
Sun, W. et al. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic acids research 37, 5365–5377 (2009).
Zhang, J. et al. A molecular inversion probe-based next-generation sequencing panel to detect germline mutations in Chinese early-onset colorectal cancer patients. Oncotarget 8, 24533–24547 (2017).
Acknowledgements
We thank the technical support of Gemma Aiza and the help of the staff of the Genetic Diagnostics and Genetic Counselling Units of the Hereditary Cancer Program at the Catalan Institute of Oncology in Hospitalet de Llobregat, Badalona, and Girona, in particular Marta Pineda, Sara González, Mireia Menéndez, Silvia Iglesias, Ares Solanes, Elia Grau, and Esther Darder. This study was funded by the Spanish Ministry of Science, Innovation and Universities, co-funded by FEDER funds- a way to build Europe, [SAF2016-80888-R (LV), SAF2015-68016-R (GC), Formación de Personal Investigador (IQ)]; Instituto de Salud Carlos III [CIBERONC CB16/12/00234, PI16/00563 (CL), Sara Borrell postdoctoral contract (PM)], the Government of Catalonia [Department of Health PERIS SLT002/16/0037, SLT002/16/00164 “Acció instrumental d’incorporació de científics i tecnòlegs” (MT)], AGAUR 2017SGR1282 and CERCA Program] and Fundación Olga Torres. This study has been enabled by COST Action CA17118.
Author information
Authors and Affiliations
Contributions
L.V. supervised the study and developed its concept and design. S.B., I.Q., P.M. and M.T. performed experiments and/or acquired data. P.M.M.-T., S.B., M.H.A. and V.M. performed the analysis of NGS, SNP array and methylation data. C.L., M.N., V.P., J.B. and G.C. contributed to the acquisition of samples and clinical data. S.B., I.Q., P.M.M.-T., P.M., M.H.A., M.T., V.M., G.C. and L.V. contributed to the analysis and interpretation of results. L.V. drafted the manuscript. All authors critically reviewed the manuscript for important intellectual content and approved its final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Belhadj, S., Quintana, I., Mur, P. et al. NTHL1 biallelic mutations seldom cause colorectal cancer, serrated polyposis or a multi-tumor phenotype, in absence of colorectal adenomas. Sci Rep 9, 9020 (2019). https://doi.org/10.1038/s41598-019-45281-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45281-1
This article is cited by
-
Genotype–Phenotype Correlations in Autosomal Dominant and Recessive APC Mutation-Negative Colorectal Adenomatous Polyposis
Digestive Diseases and Sciences (2023)
-
NTHL1 is a recessive cancer susceptibility gene
Scientific Reports (2023)
-
An integrative in-silico analysis discloses a novel molecular subset of colorectal cancer possibly eligible for immune checkpoint immunotherapy
Biology Direct (2022)
-
Intestinal and extraintestinal neoplasms in patients with NTHL1 tumor syndrome: a systematic review
Familial Cancer (2022)
-
Danish guidelines for management of non-APC-associated hereditary polyposis syndromes
Hereditary Cancer in Clinical Practice (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
