
Abstract
Interferon (IFN)-αs bind to and activate their cognate cell surface receptor to invoke an antiviral response in target cells. Well-described receptor-mediated signaling events result in transcriptional regulation of IFN sensitive genes, effectors of this antiviral response. Results from a pilot study to evaluate the clinical efficacy of IFN-α treatment of SARS patients provided evidence for IFN-inducible resolution of disease. In this report we examined the contribution of IFN-inducible phosphorylation-activation of specific signaling effectors to protection from infection by a SARS-related murine coronavirus, MHV-1. As anticipated, the earliest receptor-activation event, Jak1 phosphorylation, is critical for IFN-inducible protection from MHV-1 infection. Additionally, we provide evidence for the contribution of two kinases, the MAP kinase p38MAPK, and protein kinase C (PKC) δ to antiviral protection from MHV-1 infection. Notably, our data suggest that MHV-1 infection, as for the Urbani SARS coronoavirus, inhibits an IFN response, inferred from the lack of activation of pkr and 2′5′-oas, genes associated with mediating the antiviral activities of IFN-αs. To identify potential target genes that are activated downstream of the IFN-inducible signaling effectors we identified, and that mediate protection from coronavirus infection, we examined the gene expression profiles in the peripheral blood mononuclear cells of SARS patients who received IFN treatment. A subset of differentially regulated genes were distinguished with functional properties associated with antimicrobial activities.
Similar content being viewed by others


Introduction
In March 2003, a number of laboratories independently reported the isolation and characterization of a novel coronavirus (CoV) from specimens of patients with severe acute respiratory syndrome (SARS) 1, 2. Although an effective therapeutic strategy against SARS has yet to be developed, interferons (IFN) are recognized to play critical roles in host resistance to viral infection 3, 4, 5, 6, thereby implicating them as potential candidates for SARS CoV treatment. IFNs inhibit viral infection by directly interfering with viral replication and by inducing both an innate and adaptive immune response to infection. IFN-αs are effective in the treatment of hepatitis B and C 7, 8 and respiratory coronavirus infections unrelated to the Urbani strain of SARS-CoV 9, 10, 11, 12. IFN-αs and IFN-β 13, 14, 15, 16, 17, and combinations of IFN-α/β and IFN-γ 15, 18 are effective in inhibiting SARS-CoV replication in vitro. Moreover, it was reported that macacque monkeys were protected from infection with SARS CoV by treatment with IFN-α 19.
IFN alfacon-1 (Infergen, Intermune Corp, Brisbane, California), a synthetic IFN-α designed to represent a consensus IFN-α 20, has been shown in both cell culture systems 21 and comparative clinical trials 22 to inhibit viral replication more potently than other IFN-αs. Based on these findings, a pilot study to evaluate the potential clinical benefit and safety of IFN alfacon-1 in SARS treatment was conducted by our laboratory. Patients treated with IFN alfacon-1 and corticosteroids had a shorter time to 50% resolution of lung radiographic abnormalities, better oxygen saturation, and resolved their need for supplemental oxygen more rapidly, when compared with a group receiving corticosteroids alone 23.
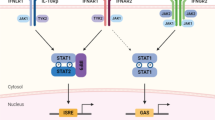
IFN-αs and IFN-β bind to and activate the Type I IFN receptor, IFNAR 24. High affinity binding of IFNs to IFNAR induces association of the two receptor chains and subsequent reciprocal transphosphorylation and activation of the associated Janus kinases (Jaks). The Jak-STAT (signal transducer and activator transcription) pathway induces IFN-mediated signal transduction and transcriptional activation, and is required for antiviral responses 25. In recent years it has become apparent that activation of the IFNR-associated Jaks also regulates multiple downstream signal transducing molecules, such as the p38 mitogen activated protein kinase (MAPK) 26, CrkL 27, phosphatidylinositol 3 kinase 28, the protein kinase C family (PKC) 29, and the Vav proto-oncogene 30. Such signaling diversity is consistent with the pleiotropic biological effects of the IFNs in target cells and tissues 31.
Mouse Hepatitis Viruses (MHV) are type 2 coronaviruses that typically cause pneumonitis, encephalitis, entercolitis and hepatitis. 32, 33, 34. Phylogenetic data suggest that the SARS-CoV is a distinct group 2 coronavirus 35, 36. Previous studies have shown that IFNs-α/β provide protection from MHV-A59 and MHV-2 infections in vivo 37, 38. In ongoing experiments, we have evidence that MHV-1, when injected intra-nasally into mice, will induce a lung infection indistinguishable from that of the SARS-CoV infection in humans (G Levy et al., UHN, unpublished). Specifically, A/J mice are highly susceptible to intranasal infection with MHV-1, develop pulomonary disease characterized by marked pulmonary congestion, hyaline membranes, interstitial thickening and inflammation and alveolar exudates similar to SARS lung disease in humans. Accordingly, to understand the mechanism of action of IFN alfacon-1 in limiting SARS-CoV infection in humans, we developed an in vitro model system, using C3H murine L2 lung fibroblast cells, permissive for MHV-1 infection. In this report we provide evidence for the direct antiviral effects of IFN-α against MHV-1 and identify several signaling effectors which mediate these effects. Additionally, by examining the changes in gene expression profiles in the PBMC of SARS patients who received IFN alfacon-1 treatment, we identify a subset of IFN-responsive genes whose differential expression may influence resolution of disease.
Materials and Methods
Cells, virus and reagents
L2, C3H murine lung fibroblast cells 39 were maintained in DMEM/ 10% heat inactivated fetal calf serum with 100U/ml penicillin, 100mg/ml streptomycin, 4.5 g/L L-glutamine. Murine IFN-α4 was provided by Dirk Gewert (Wellcome Research Lab, Kent, UK). The pharmacological inhibitors SB203580, Rottlerin, Jak inhibitor 1, and AG490 were obtained from Calbiochem. Polyclonal Abs against phosphorylated STAT1 (Tyr701), p38 MAPK (Thr180/Tyr182), PKCδ (Thr505), and Jak1 (Tyr1022/1023) were obtained from Cell Signaling Technology, polyclonal Abs against STAT1-α p91, p38 MAPK, and PKCd were obtained from Santa Cruz Biotechnology, while the polyclonal Ab against Jak1 was from Upstate Biotechnology, and the monoclonal Ab against STAT3 was obtained from Zymed Laboratories. Stock MHV-1 at a titer of 2.9 ×105 PFU/ml was used for all experiments.
28S rRNA degradation assay
106 L2 cells were either left untreated or treated with prescribed doses of murine IFN-α4 for 14 h, then infected with MHV-1 for 36 h. Cytoplasmic RNA was isolated using the Qiagen RNeasy mini kit according to the manufacturer's protocol, and then resolved by electrophoresis in a 1% denaturing agarose-formaldehyde gel. Northern blot analysis was performed as previously described 40. A [γ-32P]ATP-labeled oligonucleotide probe (5′-CTA ATC ATT CGC TTT ACC GG-3′), which specifically binds to nucleotides -1532 to -1551 from the 5′ end of murine 28S rRNA, was used for the detection of 28S rRNA and its cleavage products.
Antiviral assays
The assay for IFN-induced antiviral activity in monolayer cells was described as previously 41, 42, 43. Briefly, 104 cells were seeded in 96-well tissue culture plates in DMEM containing 2% FCS. After 24 h, appropriate dilutions of IFN-α4 were added and cells were incubated for an additional 24 h. Then, medium containing IFN was aspirated and the appropriate dilution of MHV-1 in a volume of 200μl was added to the cells. After a further 24 h, cells were fixed in 95% ethanol, stained with crystal violet solution (0.1% in 2% ethanol) and destained in 0.5M NaCl, 50% ethanol. IFN-induced inhibition of viral replication was assessed by reading the absorbance at 570 nm using a Microplate reader (Molecular Devices) and SOFTmax® 2.32 software relative to infected, untreated cells.
Cell lysis and Western blot
For immunoblotting, L2 cells were stimulated with IFN-α4 (104 U/ml) for the indicated times and lysed in phosphorylation lysis buffer. Immunoprecipitations and immunoblotting using an ECL (enhanced chemiluminescence) method were performed as described previously 45. In experiments in which pharmacological inhibitors were used, the cells were pre-treated for 60 min with indicated concentrations of the inhibitors and subsequently treated with IFN-α4 prior to lysis in phosphorylation lysis buffer.
RNA extraction, cDNA synthesis and real-time PCR
To harvest RNA for Real Time PCR, 106 L2 cells were either left untreated, treated with MHV-1 or IFN-α4 (104U/ml) for the indicated times, or treated with pharmacological inhibitors for 1 h prior to treatment with 104U/ml IFN-α4 for 12 h. Cells were lysed and homogenized using Qiagen QIA-shredder columns and RNA isolation was performed using the Qiagen RNeasy mini kit according to the manufacturer's protocol. cDNA was synthesized using 1 mg RNA in the presence of random primers and AMV Reverse Tanscriptase for 1 h at 42°C (Promega). Reaction components were obtained from the LightCycler® FastStart DNA Master SYBR GreenPLUS I Kit (Roche). The LightCycler® instrument (Roche) and corresponding software were used for all reactions. The PCR was performed in a final volume of 20 μl, 0.5 μM of each primer and 5 ml template cDNA (concentration 100 ng/ml). Primer sets were as follows, gapdh forward primer 5′-CCT GCA CCA CCA ACT GCT TA-3′ and the reverse primer 5′-TCA TGA GCC CTT CCA CAA TG-3′, pkr forward 5′-GGC TCC TGT GTG GGA AGT CA-3′ and the reverse primer 5′-TAT GCC AAA AGC CAG AGT CCT T-3′, 2′5′oas forward primer 5′-TGA GCG CCC CCC ATC T-3′ and the reverse primer 5′-CAT GAC CCA GGA CAT CAA AGG-3′. Standard curves were established for each primer set and both reference and target reactions were performed for each sample.
Affymetrix analysis of ISG expression in PBMC of SARS patients
Serial peripheral blood mononuclear cells (PBMC) from 3 of the 22 SARS patients included in the IFN alfacon-1 pilot study 44 were analyzed for gene expression profiles using the Affymetrix U113A 2.0 plus gene chips. As described in Table 1 and 2, 2 patients received subcutaneous IFN alfacon-1 in addition to the standard of care, corticosteroid treatment. RNA was extracted from PBMC. PBMC were lysed, homogenized using Qiagen QIA-shredder columns and RNA isolation was performed using the Qiagen RNeasy mini kit according to the manufacturer's protocol. To ensure effective inactivation of RNases, lysis reagents contained denaturing guanidine isothiocyanate. A high-salt buffer system allowed RNA to selectively bind the silica-gel membrane of the spin column, while contaminants were washed away. Total cellular RNA was eluted in water. Total RNA was amplified using the Ambion MessageAmp™ II aRNA Amplification Kit. Amplification was performed according to the manufacturer's protocol. Briefly, during the first round of amplification, 10 ml (1000 ng) of total RNA was used, and converted to aRNA. aRNA concentrations were calculated, and 2 mg of amplified RNA (aRNA) was used in the second round of amplification. During second round amplification, biotin labeled NTPs (10mM biotin-11-CTP and 10mM biotin-16-UTP) were incorporated into the cRNA. 30 mg of biotinylated cRNA was sent for Affymetrix gene expression analysis to the Centre for Applied Genomics Microarray Facility at The Hospital for Sick Children, Toronto Ontario. An additional 5 μl (200 ng/μl) was sent for Bioanalyzer analysis.
Gene expression analysis was performed by hybridization of cDNA to the Affymetrix® gene chip – U133A 2.0 plus. This chip codes for over 50 000 probes. The specific effect of IFN alfacon-1 treatment on gene expression was analyzed using the microarray analysis software, Array Assist®. Each gene expression profile was analyzed using GCRMA analysis. This method uses a basic algorithm in order to identify genes whose expression is altered/regulated in the sample array when compared to the basal levels expressed on the Affymetrix® chip. From this list of genes, using the gene expression levels measured on day 6 relative to onset of symptoms, designated as 'basal' for each SARS patient, a new list of genes whose expression was regulated greater than three fold by IFN treatment was obtained.
Results
The data in panels A and B in Figure 1 show IFN-α4-dependent protection of L2 cells from MHV-1-inducible 28S RNA cleavage and cytopathic effects (CPE). To examine the influence of specific IFN activated signaling effectors in protection from MHV-1 infection, we studied the effects of a number of pharmacological inhibitors targeted against specific signaling molecules. High affinity binding of IFN to IFNAR leads to the phosphorylation-activation of the receptor-associated Jaks, Jak1 and Tyk2. L2 cells treated with the Jak1 inhibitor, Jak inhibitor 1, led to a reduction in IFN-α4 induced tyrosine phosphorylation of Jak1 (Figure 1C). Additionally, we observed a reduction in IFN-inducible tyrosine phosphorylation of STAT1, STAT2 and STAT3 (Figure 1D). When L2 cells were treated with the Jak 1 inhibitor prior to IFN treatment, then infected with MHV-1, we observed a reduction in IFN-inducible protection from MHV-1 infection (Figure 1B). As a negative control, we examined the effect of the inhibitor AG490, which specifically inhibits Jak2 at 100 μM 45. As anticipated, treating cells with 100 μM AG490 did not influence the anti-MHV-1 activity of IFN-α4 (Figure 1B).

The pharmacological inhibitors: Jak inhibitor 1, Rottlerin and SB203580 inhibit IFN-inducible anti-MHV-1 activity. (A) 106 L2 cells were either left untreated, or treated with the indicated doses of IFN-α4 for 14 h, then challenged with MHV-1 for 36 h. Cells were lysed, RNA extracted and resolved by electrophoresis in agarose and Northern analyses of 28S RNA and cleavage products performed. Data are representative of 2 independent experiments. (B) 104 L2 cells were either left untreated (▴) or treated with 10μM Jak inhibitor 1(▪), 1μM Rottlerin (□), 10μM SB203580 (▵), or 100μM AG490 (O) for 1 h. Cells were then incubated, in triplicate, with the indicated dose of IFN-α4 for 16 h, then challenged with MHV-1. After 24 h CPE were quantitated using a colorimetric assay (Materials and Methods). Data are expressed as percent protection from CPE. Mean values ± SE of three independent experiments are shown. (C) 107 L2 cells were either left untreated, or treated with 10μM Jak Inhibitor 1, 10 μM SB203580 or 1μM Rottlerin for 1 h prior to a 30 min IFN-α4 (104 U/ml) treatment. Cells were lysed and equal amounts of whole cell lysates were resolved by SDS-PAGE and examined for Jak1, p38 MAPK or PKCδ phosphorylation by immunoblotting with anti-phospho-Jak1,-p38 MAPK or -PKCδ antibodies. Membranes were stripped and re-probed with anti-Jak1, anti-p38 MAPK, or anti-PKCδ antibodies to control for loading. Blots shown are representative of 3 independent experiments. (D) 107 L2 cells were either left untreated, or treated with 10 μM Jak Inhibitor 1 or 1 μM Rottlerin for 1 h prior to a 30 min IFN-α4 (104 U/ml) treatment. Cells were lysed and equal amounts of whole cell lysates were resolved by SDS-PAGE and examined for STAT tyrosine phosphorylation by immunoblotting with anti-phospho-STAT antibodies. Membranes were stripped and re-probed with anti-STAT antibodies to control for loading. Blots shown are representative of 3 independent experiments.
The p38 MAPK pathway is essential for induction of antiviral responses by IFNs against hepatitis C virus 46, vesicular stomatitis virus and encephalomyocarditis virus infections 47, 48. To examine the importance of IFN-inducible p38 MAPK signaling in protection against MHV-1 infection, L2 cells were treated with SB203580 prior to IFN-α4. 10 μM SB203580 reduced the IFN-α4-induced phosphorylation of p38 MAPK (Figure 1C) and IFN-inducible protection from MHV-1 infection (Figure 1B). In subsequent experiments, using the PKCδ inhibitor Rottlerin, we studied the influence of PKCδ on IFN-inducible anti-MHV-1 activity. 1 μM Rottlerin decreased the IFN-α4 induced tyrosine phosphorylation of PKCδ, (Figure 1C). Additionally, Rottlerin reduced the anti-MHV-1 activity of IFN-α4 (Figure 1B). As expected, inhibition of PKCδ did not decrease the IFN-α4 induced tyrosine phosphorylation of STAT1 (Figure 1D), since inhibition of PKCδ activity blocks Ser727 STAT1 phosphorylation 29.
IFN-inducible control of viral replication is effectively regulated by the transcriptional activation of key interferon stimulated genes (ISG) such as the RNA-dependent protein kinase (PKR) and 2′5′- oligoadenylate synthetase (OAS)/ endoribonuclease L (RNase L) 49, 50. In murine cells, virus-inducible activation of Toll like receptors leads to the transcriptional activation of IFN-β, and IFN-α4 51, 52. Therefore, MHV-1 infection per se should induce transcriptional activation of IFNs, and lead to the subsequent transcriptional activation of pkr and 2′5′-oas. Accordingly, L2 cells were infected with MHV-1, and RNA harvested at different time points. The extent of gene expression was analyzed using real time PCR. In time course studies we observed minimal transcriptional activation for both pkr and 2′5′-oas by MHV-1 (Figure 2A). In subsequent experiments we examined the effect of IFN-α4 treatment on their transcriptional activation. In a similar time course study, when L2 cells were treated with 104 U/ml IFN-α4, transcriptional activation of both 2′5′-oas and pkr were observed (Figure 2B).

MHV-1 infection leads to negligible transcriptional activation of 2′5′-oas and pkr. 106 L2 cells were either left untreated (UT), infected with MHV-1 (A), treated with 104U/ml IFN-α4 for the times indicated (B), or treated with 10 μM SB203580, 1 μM Rottlerin or 10μM Jak 1 inhibitor for 1h then 104U/ml IFN-α4 for 12h (C). RNA was extracted, cDNA synthesized and quantitative real-time PCR analyses performed for reference GAPDH and target genes 2′5′-oas and pkr expression. Data are presented as the fold-change in expression compared to untreated L2 cells. Values ± SE were calculated using Relative Quantification software (Roche) and are the mean of three separate reactions, each performed in triplicate.
In subsequent experiments we examined the effects of inhibiting IFN-α4 inducible activation of Jak1, p38 MAPK and PKCδ on IFN-α4 inducible transcriptional activation of pkr and 2′5′-oas gene expression. L2 cells were treated with 10 μM Jak inhibitor 1, 10 μM SB203580 or 1 mM Rottlerin, for 1 h, then with 104 U/ml IFN-α4 for 12 h. RNA was harvested, and gene expression for pkr and 2′5′-oas examined. This 12 h time point was selected, based on the data from Figure 2B, namely that maximal IFN-inducible transcriptional activation of these genes is attained at 12h. Notably, inhibition of IFN-inducible activation of Jak1, p38 MAPK or PKCδ effectively reduced the IFN-inducible gene expression for pkr and 2′5′-oas (Figure 2C).
In the absence of any clear cause and effect relationship between pkr and 2′5′-oas gene expression and IFN-inducible anti-MHV-1 activity, we subsequently examined the signature patterns of gene expression in the PBMC from 2 of the SARS study patients we had treated with IFN alfacon-1, during the Toronto 2003 outbreak 44. For comparison we measured gene expression in the PBMC of a SARS patient that was treated with corticosteroids alone (see Materials and Methods). PBMC were sampled on days 6, 8, 10 and 13 post-onset of symptoms. For the IFN-treated SARS patients, the day 6 PBMC sample represented a baseline in the absence of IFN treatment, whereas days 8, 10 and 13 corresponded to days 2, 4 and 7 relative to the start of IFN treatment. The data in Table 3 reveal 2 lists of genes expressed differentially between the 2 IFN-treated SARS patients and the 1 non-IFN treated patient, using a cut-off of ≥ (A) or ≤ (B) 3-fold change in expression, relative to basal expression levels on day 6 post-onset of symptoms. In Figure 3, differentially expressed genes are clustered to indicate high and low gene expression, using a red-green colour scale to show the relative range. Notably, our analyses identified 11 genes that were upregulated and 21 genes that were downregulated in the IFN-treated patients, according to these criteria. Upregulated genes included interleukin 8 (IL-8) and defensins alpha 1 and alpha 4. IL-8 is a chemoattractant for neutrophils, induces neutrophil degranulation and enhances defensin release from neutrophils. Defensins are potent antimicrobial peptides that have been shown to exhibit antiviral activity against enveloped viruses and HIV 53, 54. Downregulated genes included protein-kinase repressor (PRKRIR), which acts to prevent PKR synthesis 55. Expression for exportin 1 (CRM1) was also downregulated in the PBMC of the IFN-treated SARS patients. CRM1 is a transport receptor that mediates nuclear export of proteins. CRM1 interacts with the Rev protein of HIV 56, suggestive of a potential for inhibition of nuclear activities associated with viral replication. Additionally, the expression of genes encoding RNA binding proteins and helicases including ELAV1, RNU22, DDX1 and DHX15 was also downregulated, in further support of inhibitory effects on viral replication intermediates. Interestingly, DDX1, a DEAD box protein, has been implicated in facilitating HIV replication57, 58. Viewed altogether, the implications are that IFN alfacon-1 treatment affected gene expression in the PBMC of SARS patients to promote direct antiviral effects and inhibit viral replication.

Heat map of genes regulated by IFN alfacon-1 in PBMC of SARS patients. The relative expression levels of the genes identified in Table 3 are described. Upregulated genes (A) are shown in red and downregulated genes (B) are shown in green. Maximum intensity values +/− 9 are shown. Patient 1, SARS patient treated with corticosteroids alone; Patients 2 and 3, SARS patients treated with IFN alfacon-1 (as per Tables 1 and 2).
Discussion
Using murine IFN-α4 and an in vitro model for SARS, namely MHV-1 infection of murine lung fibroblast cells, we investigated the influence of various signaling molecules during IFN-α4 mediated antiviral protection. Our findings suggest that Jak1, PKCδ, and p38 MAPK play important roles in IFN-α-mediated anti-MHV-1 activity. At the lower doses of IFN-α4, pre-treating the cells with Jak 1 inhibitor completely abrogated the protective effects of IFN-α4. Since Jak1 activation is an early post-receptor activation event, it is not surprising that abrogating this upstream effector in the IFN-receptor signaling cascade has a profound effect on subsequent downstream signaling events and biological responses. By contrast, at the higher doses of IFN-α4, inhibition of PKCδ inhibited IFN-induced antiviral activity to a greater extent, implying that STAT1-sensitive ISG activation is necessary for IFN-inducible anti-MHV-1 activity. Notably, IFN-α4-induced antiviral activity is unlikely to be exclusively attributable to the activities of Jak1, p38 MAPK, and PKCδ.
Further, we demonstrate that L2 cells treated with IFN-α4 induce the transcriptional activation of pkr and 2′5′-oas, in stark contrast to the negligible activation observed when cells were infected with MHV-1. We infer from these data that MHV-1 infection per se may prevent the activation of the IFN system. Indeed, many different viruses evade an immune response by interfering with transcriptional activation of IFN-α/β genes 59. Notably, in order to escape activation of the IFN system, the SARS-CoV appears to block a step after the early nuclear transport of IRF-3, the transcription factor essential for IFN-β and IFN-α4 promoter activity 60.
Inhibition of IFN-induced activation of Jak1, PKCδ and p38 MAPK was shown to also influence the transcriptional activation of pkr and 2′5′-oas. The anti-MHV-1 effects of IFN-α4 are unlikely to be wholly attributable to the inhibitory activities of PKR and 2′5′OAS. Indeed, many other IFN-inducible genes have been identified which encode factors implicated in IFN-induced antiviral responses 50, 61, 62, 63. Interestingly, when we analyzed the signature patterns of gene expression in the PBMC of 2 SARS patients that received IFN alfacon-1, compared with a patient that did not, we observed a trend of differential gene expression in the IFN-treated patients that was suggestive of antimicrobial activities. In ongoing studies we are investigating the contribution of their specific gene products to antiviral activity.
In conclusion, our data suggest that activation of Jak1, p38 MAPK and PKCδ are involved in the generation of IFN-α-mediated anti-MHV-1 activity, and in the control of IFN-induced transcriptional activation of, minimally, pkr and 2′5′-oas. The implications are that IFN-inducible activation of specific signaling molecules leads to the transcriptional regulation of a subset of ISGs that determine the direct antiviral activity of IFN-α against coronavirus infections.
References
Rota PA, Oberste MS, Monroe SS, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003; 300:1394–9.
Poutanen SM, Low DE, Henry B, et al. Identification of severe acute respiratory syndrome in Canada. N Engl J Med 2003; 348:1995–2005.
Brierley MM, Fish EN . Review: IFN-α/β receptor interactions to biologic outcomes: understanding the circuitry. J Interferon Cytokine Res 2002; 22:835–45.
Samuel CE . Antiviral actions of interferons. Clin Microbiol Rev 2001; 14:778–809, table of contents.
Levy DE, Garcia-Sastre A . The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev 2001; 12:143–56.
Biron CA . Interferons alpha and beta as immune regulators-a new look. Immunity 2001; 14:661–4.
Nguyen MH, Wright TL . Therapeutic advances in the management of hepatitis B and hepatitis C. Curr Opin Infect Dis 2001; 14:593–601.
Cornberg M, Wedemeyer H, Manns MP . Hepatitis C: therapeutic perspectives. Forum (Genova) 2001; 11:154–62.
Higgins PG, Phillpotts RJ, Scott GM, et al. Intranasal interferon as protection against experimental respiratory coronavirus infection in volunteers. Antimicrob Agents Chemother 1983; 24:713–5.
Tyrrell DA . The efficacy and tolerance of intranasal interferons: studies at the Common Cold Unit. J Antimicrob Chemother 1986; 18 Suppl B:153–6.
Turner RB, Felton A, Kosak K, et al. Prevention of experimental coronavirus colds with intranasal alpha-2b interferon. J Infect Dis 1986; 154:443–7.
Sperber SJ, Hayden FG . Comparative susceptibility of respiratory viruses to recombinant interferons-alpha 2b and -beta. J Interferon Res 1989; 9:285–93.
Cinatl J, Morgenstern B, Bauer G, et al. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 2003; 361:2045–6.
Stroher U, DiCaro A, Li Y, et al. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon- alpha. J Infect Dis 2004; 189:1164–7.
Scagnolari C, Vicenzi E, Bellomi F, et al. Increased sensitivity of SARS-coronavirus to a combination of human type I and type II interferons. Antivir Ther 2004; 9:1003–11.
Zheng B, He ML, Wong KL, et al. Potent inhibition of SARS-associated coronavirus (SCOV) infection and replication by type I interferons (IFN-alpha/beta) but not by type II interferon (IFN-gamma). J Interferon Cytokine Res 2004; 24:388–90.
Hensley LE, Fritz LE, Jahrling PB, et al. Interferon-beta 1a and SARS coronavirus replication. Emerg Infect Dis 2004; 10:317–9.
Sainz B, Jr., Mossel EC, Peters CJ, Garry RF . Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV). Virology 2004; 329:11–7.
Haagmans BL, Kuiken T, Martina BE, et al. Pegylated interferon-alpha protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat Med 2004; 10:290–3.
Alton K Stabinsky Y, Richards R, et al. Production, characterization and biological effects of recombinant DNA derived humans IFN-alfa and IFN-gamma analogs. DeMaeyer and H. Schellekens (eds.) Amsterdam, the Netherlands: Elsevier Science 1983:119–28
Blatt LM, Davis JM, Klein SB, Taylor MW . The biologic activity and molecular characterization of a novel synthetic interferon-alpha species, consensus interferon. J Interferon Cytokine Res 1996; 16:489–99.
Melian EB, Plosker GL . Interferon alfacon-1: a review of its pharmacology and therapeutic efficacy in the treatment of chronic hepatitis C. Drugs 2001; 61:1661–91.
Loutfy MR, Blatt LM, Siminovitch KA, et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. Jama 2003; 290:3222–8.
Pestka S, Langer JA, Zoon KC, Samuel CE . Interferons and their actions. Annu Rev Biochem 1987; 56:727–77.
Stark GR, Kerr IM, Williams BR, et al. How cells respond to interferons. Annu Rev Biochem 1998; 67:227–64.
Uddin S, Majchrzak B, Woodson J, et al. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem 1999; 274:30127–31.
Fish EN, Uddin S, Korkmaz M, et al. Activation of a CrkL-stat5 signaling complex by type I interferons. J Biol Chem 1999; 274:571–3.
Uddin S, Fish EN, Sher DA, et al. Activation of the phosphatidylinositol 3-kinase serine kinase by IFN-alpha. J Immunol 1997; 158:2390–7.
Uddin S, Sassano A, Deb DK, et al. Protein kinase C-delta (PKC-delta ) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J Biol Chem 2002; 277:14408–16.
Platanias LC, Sweet ME . Interferon alpha induces rapid tyrosine phosphorylation of the vav proto-oncogene product in hematopoietic cells. J Biol Chem 1994; 269:3143–6.
Platanias LC . Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 2005; 5:375–86.
Sabesin SM, Koff RS . Pathogenesis of experimental viral hepatitis (second of two parts). N Engl J Med 1974; 290:996–1002.
Lavi E, Gilden DH, Highkin MK, Weiss SR . The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab Anim Sci 1986; 36:130–5.
Matthews AE, Weiss SR, Paterson Y . Murine hepatitis virus—a model for virus-induced CNS demyelination. J Neurovirol 2002; 8:76–85.
Snijder EJ, Bredenbeek PJ, Dobbe JC, et al. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol 2003; 331:991–1004.
Stadler K, Masignani V, Eickmann M, et al. SARS—beginning to understand a new virus. Nat Rev Microbiol 2003; 1:209–18.
de Haan CA, de Wit M, Kuo L, et al. The glycosylation status of the murine hepatitis coronavirus M protein affects the interferogenic capacity of the virus in vitro and its ability to replicate in the liver but not the brain. Virology 2003; 312:395–406.
Matsuyama S, Henmi S, Ichihara N, et al. Protective effects of murine recombinant interferon-beta administered by intravenous, intramuscular or subcutaneous route on mouse hepatitis virus infection. Antiviral Res 2000; 47:131–7.
Sturman LS, Takemoto KK . Enhanced growth of a murine coronavirus in transformed mouse cells. Infect Immun 1972; 6:501–7.
Banerjee S, An S, Zhou A, et al. RNase L-independent specific 28S rRNA cleavage in murine coronavirus-infected cells. J Virol 2000; 74:8793–802.
Fish EN, Hannigan GE, Banerjee K, Williams BR . The interaction of interferon-alpha and -gamma: regulation of (2–5)A synthetase activity. Virology 1988; 165:87–94.
Kerr IM, Stark GR . The antiviral effects of the interferons and their inhibition. J Interferon Res 1992; 12:237–40.
Dahl H, Degre M . Separation of antiviral activity of human interferon from cell growth inhibitory effect. Nature 1975; 257:799–800.
MR Loutfy LB, KA Siminovitch, S Ward, et al. Interferon alfacon-1 plus corticosteroids in sever acute respiratory syndrome. JAMA 2003; 290:3222–8.
Nielsen M, Kaltoft K, Nordahl M, et al. Constitutive activation of a slowly migrating isoform of Stat3 in mycosis fungoides: tyrphostin AG490 inhibits Stat3 activation and growth of mycosis fungoides tumor cell lines. Proc Natl Acad Sci U S A 1997; 94:6764–9.
Ishida H, Ohkawa K, Hosui A, et al. Involvement of p38 signaling pathway in interferon-alpha-mediated antiviral activity toward hepatitis C virus. Biochem Biophys Res Commun 2004; 321:722–7.
Goh KC, Haque SJ, Williams BR . p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. Embo J 1999; 18:5601–8.
Mayer IA, Verma A, Grumbach IM, et al. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABL-expressing cells. J Biol Chem 2001; 276:28570–7.
Khabar KS, Dhalla M, Siddiqui Y, et al. Effect of deficiency of the double-stranded RNA-dependent protein kinase, PKR, on antiviral resistance in the presence or absence of ribonuclease L: HSV-1 replication is particularly sensitive to deficiency of the major IFN-mediated enzymes. J Interferon Cytokine Res 2000; 20:653–9.
M.M. Brierley ENF. IFN-a/b receptor interactions to biologic outcomes: understanding the circuitry. J IFN Cytokine Res 2002; 22:835–845.
Kawai T, Sato S, Ishii KJ, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 2004; 5:1061–8.
Malmgaard L . Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res 2004; 24:439–54.
Chang TL, Vargas J, Jr., DelPortillo A, Klotman ME . Dual role of alpha-defensin-1 in anti-HIV-1 innate immunity. J Clin Invest 2005; 115:765–73.
Zhang L, Yu W, He T, et al. Contribution of human alpha-defensin 1, 2, and 3 to the anti-HIV-1 activity of CD8 antiviral factor. Science 2002; 298:995–1000.
Gale M, Jr., Blakely CM, Hopkins DA, et al. Regulation of interferon-induced protein kinase PKR: modulation of P58IPK inhibitory function by a novel protein, P52rIPK. Mol Cell Biol 1998; 18:859–71.
Fontoura BM, Faria PA, Nussenzveig DR . Viral interactions with the nuclear transport machinery: discovering and disrupting pathways. IUBMB Life 2005; 57:65–72.
Fang J, Kubota S, Yang B, et al. A DEAD box protein facilitates HIV-1 replication as a cellular co-factor of Rev. Virology 2004; 330:471–80.
Krishnan V, Zeichner SL . Alterations in the expression of DEAD-box and other RNA binding proteins during HIV-1 replication. Retrovirology 2004; 1:42.
Conzelmann KK . Transcriptional activation of alpha/beta interferon genes: interference by nonsegmented negative-strand RNA viruses. Journal of Virology 2005; 79:5241–8.
Spiegel M, Pichlmair A, Martinez-Sobrido L, et al. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J Virol 2005; 79:2079–86.
Der SD, Zhou A, Williams BR, Silverman RH . Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 1998; 95:15623–8.
Geiss G, Jin G, Guo J, et al. A comprehensive view of regulation of gene expression by double-stranded RNA-mediated cell signaling. J Biol Chem 2001; 276:30178–82.
Taylor MW, Grosse WM, Schaley JE, et al. Global effect of PEG-IFN-alpha and ribavirin on gene expression in PBMC in vitro. J Interferon Cytokine Res 2004; 24:107–18.
Acknowledgements
The authors are grateful to Ms Beata Majchrzak-Kita for technical assistance and to Mr Ramtin Rahbar for his critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zorzitto, J., Galligan, C., Ueng, J. et al. Characterization of the antiviral effects of interferon-α against a SARS-like coronoavirus infection in vitro. Cell Res 16, 220–229 (2006). https://doi.org/10.1038/sj.cr.7310030
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310030
Keywords
This article is cited by
-
Chemokine CCL6 Plays Key Role in the Inhibitory Effect of Vitamin A on Norovirus Infection
Journal of Microbiology (2023)
-
The COVID-19 pandemic: biological evolution, treatment options and consequences
Innovative Infrastructure Solutions (2020)
-
What are the drugs having potential against COVID-19?
Medicinal Chemistry Research (2020)
