
Abstract
The genomes of eukaryotic cells are under continuous assault by environmental agents and endogenous metabolic byproducts. Damage induced in DNA usually leads to a cascade of cellular events, the DNA damage response. Failure of the DNA damage response can lead to development of malignancy by reducing the efficiency and fidelity of DNA repair. The NBS1 protein is a component of the MRE11/RAD50/NBS1 complex (MRN) that plays a critical role in the cellular response to DNA damage and the maintenance of chromosomal integrity. Mutations in the NBS1 gene are responsible for Nijmegen breakage syndrome (NBS), a hereditary disorder that imparts an increased predisposition to development of malignancy. The phenotypic characteristics of cells isolated from NBS patients point to a deficiency in the repair of DNA double strand breaks. Here, we review the current knowledge of the role of NBS1 in the DNA damage response. Emphasis is placed on the role of NBS1 in the DNA double strand repair, modulation of the DNA damage sensing and signaling, cell cycle checkpoint control and maintenance of telomere stability.
Similar content being viewed by others

Introduction
The genomes of eukaryotic cells are under continuous assault by environmental agents, including ultraviolet light, ionizing radiation, and reactive chemicals, as well as the byproducts of normal intracellular metabolism including reactive oxygen intermediates and inaccurately replicated DNA 1. One of the most dangerous DNA lesions is the DNA double strand break (DSB), which usually leads to a cascade of cellular events, termed the DNA damage response. The DNA damage response involves sensing DNA damage, transduction of the damage signal to a network of cellular pathways, including cell cycle checkpoints, DNA repair, stress responses to telomere maintenance, and apoptosis, etc. 2 (Figure 1). Failure in the DNA damage response affect the level of cell survival, increase the frequency of gene mutation or chromosomal instability, and other cellular phenotypic abnormalities, which are the important mechanisms of carcinogenesis.

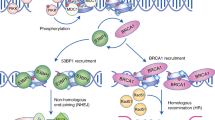
The DNA damage response network. The DNA damage such as DSB was detected by sensor(s). The sensor(s) then triggers the activation of a transducer system, which usually are a protein kinase cascade. The activated transducer(s) will amplify and diversify the signal by targeting a set of down stream effector(s), which leads to cellular events, such as DNA repair, cell cycle control, telomere stability maintenance, apoptosis, etc. Multiple sensors, transducers, and effectors participate in this cellular response. The final consequence of DNA damage in cells will be determined by types and magnitude of stress and the appropriateness of cellular response.
The NBS1 gene contains 16 exons encompassing 48797 bps of genomic sequence on chromosome 8q21 3, 4, 5, 6, 7. The NBS1 protein is a component of the MRE11/RAD50/NBS1 complex (MRN) that plays a critical role in the cellular response to DNA damage and the maintenance of chromosomal integrity. The complete disruption of NBS1 in mice is lethal 8, 9. The hypomorphic mutations which lead to decreased function of the NBS1 gene are responsible for Nijmegen breakage syndrome (NBS), a rare autosomal recessive hereditary disorder that imparts an increased predisposition to the development of malignancy 10, 11, 12. In the Polish NBS registry, 18 of 48 patients had developed lymphomas by the age of 15 13. Other types of cancers are also reported in the NBS1 compromised population 14. Cells isolated from NBS patients exhibit cellular phenotypic alterations, very similar to those seen in ataxia telangectasia (AT) that is caused by the mutation of ataxia telangectasia mutated (ATM) gene; these alterations include hypersensitivity to ionizing radiation (IR), chromosomal fragility, and abnormal cell cycle checkpoint regulation 3, 9, 15. The down-regulation of NBS1 protein level by small interfering RNA (siRNA) leads to an increase in IR-induced mutation frequency in human lymphoblastoid cells 16. Even though the severest form of NBS is recessive and of low frequency in the population, the incidence of NBS heterozygotes is estimated to be 1/177 in some southeastern areas in Europe 17. Considering the report that mice heterozygous for NBN (the murine homologue of NBS1) developed a wide variety of tumors affecting the liver, mammary gland, prostate, and lung, in addition to lymphomas 8, the NBS1 partial deficiency in human could be a considerable threat to public health.
The human NBS1 protein is composed of three functional regions at the N-terminus, central region, and the C-terminus. The N-terminus contains a forkhead associated (FHA) and breast cancer C-terminus (BRCT) domain. The FHA/BRCT domain directly binds to the histone γ-H2AX, the phosphorylated form of H2AX as a result of the presence of DSBs, then recruits other members of the MRN complex (MRE11 and RAD50) to the proximity of DSB sites 18, 19. The C-terminal region of NBS1 contains an MRE11 binding site 20, 21. As mentioned above, null mutation of NBS1 is lethal, while the mice expressing C-terminal NBS1 survive to adulthood and exhibit many phenotypes similar to NBS patients 21, 22. Consistent with that, the majority of NBS patients (homozygous 657del5) with the presence of the C-terminal NBS1 protein produced by this mutation are viable, which suggests that the maintenance of the MRE11 binding domain is essential for life 8, 23. Several serine/glutamine motifs, consensus sequences of phosphorylation by ATM and ATM/RAD3-related (ATR), are found at the central region of NBS1. In particular, the serine residues at 278 and 343 are phosphorylated by ATM kinase in response to ionizing radiation both in vitro and in vivo, and such phosphorylation is responsible for intra-S phase checkpoint control 14, 15 and telomere maintenance 11, 13, 16.
Roles in DNA DSB repair
There are at least two major pathways to repair DSBs, termed non-homologous end joining (NHEJ) and homologous recombination (HR) 24, 25. NHEJ is facilitated by DNA-dependent protein kinases (DNA-PK), composed of a catalytic subunit, DNA-PKcs, and the heterodimeric DNA binding regulatory complex Ku, which is composed of Ku70 and Ku80. The Ku heterodimer recognizes and binds the free DSB ends. DNA-PKcs is recruited and activated by the Ku complex. Artemis, a nuclease with endo- and exo-nuclease activity, is then recruited to the damage site via a direct interaction with DNA-PKcs and primes the site of damage. The DNA ligase IV/XRCC4 complex processes the DNA ends and initiates re-joining 26. In HR, DNA ends are processed to produce 3-single strand tails, and then this single strand DNA invades the intact homologous chromosome (or another homologous intra-chromosomal sequence) with the aid of RAD51. After DNA synthesis, the Holliday structure is resolved, and DNA repair is completed 19.
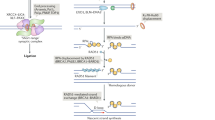
The exact function of NBS1 in DNA repair is not fully understood. NBS1 protein itself does not have DNA binding and kinase activity, which are usually required in DNA repair and DNA damage signal transduction. However, the FHA/BRCT domain at its N-terminus binds to γH2AX, and this could be an important step to recruit the MRN complex to the proximity of DNA DSB. The other two components of MRN, MRE11 and RAD50 are usually located in the cytoplasm but relocate to nuclei following DSB inducing agents; this process is abolished in the absence of NBS13. MRE11 possesses several biochemical properties which are required for DNA DSB repair, such as 3′-5′ double-strand DNA exonuclease, single-strand DNA endonuclease, and DNA unwinding activity 27, 28. RAD50 is a member of the structure maintenance of chromosome (SMC) protein family; it has two ATPase motifs at its N- and C-terminal ends and forms an anti-parallel homodimer with a flexible hinge region that may adopt a “V-like” conformation 29. The “V-like” structure could serve as a bridge to hold together the broken ends of DSB and prevent them from floating away, as well as restrict the extent of nucleotide degradation of MRE11 to prevent excessive degradation (Figure 2) 3.

The schematic architecture of MRN complex. The triple MRN complex is composed of a dimers of RAD50 and MRE11, as well as a single NBS1 28, 29, 114. MRE11 and RAD50 are recruited to the proximity of DSB site by NBS1 through the interaction of histone gH2AX with the FHA/BRCT domain in the N-terminus of NBS1 (not shown). The RAD50 dimer binds to DNA duplex with its ATPase motifs and hold the broken ends together with its coiled-coil arms 28. MRE11 associates with RAD50 and DNA duplex to prime broken ends with its nuclease activity. The MRE11 nuclease activity is modulated by NBS1 and RAD50.
In Schizosaccharomyces cerevisiae, MRE11/RAD50/XRS2, a homologue for MRN, has been reported to be involved in both pathways 30, 31, 32. The disruption of XRS2 leads to a deficiency in a special case of NHEJ termed V(D)J recombination, which is an important process during the development of the immune system to assemble a diverse array of immunoglobulin molecules and T-cell receptors 33. XRS2 mutants show hypersensitivity to IR due to insufficient NHEJ and HR activity, as well as deficiency in meiotic recombination 3, 34.
In mammalian cells, by using a cell-free DNA fragment end joining assay in vitro, the addition of human Ku, DNA-PKcs, and Ligase IV/XRCC4 complex did not promote efficient joining of cohesive-ended DNA fragments to the level observed in crude human nuclear extract. However, the addition of a cell extract fraction, which contains MRN complex, restored the DNA fragment end-joining activity to the level of the crude nuclear extract; suggesting a role of MRN in the process of DNA end-joining in human cells 35. By using the similar assays as in 35, MRN was found to promote DNA fragment end joining activity in vitro via DNA ligase IV/XRCC4 complex and DNA-PKcs is not required for this pathway 36. However, in chicken DT40 cells with a targeted disruption of the NBS1 gene, the NBS1 deficient cells showed indistinguishable NHEJ events from those wild-type cells, as measured by a plasmid DNA end joining assay in vivo 9. This is consistent with the observation that in cells from NBS patient carrying a mutated NBS1 gene, the V(D)J recombination frequencies and the quality of signal and coding joining are comparable to wild-type controls 37. The role of NBS1 in NHEJ is rather controversial when considering results obtained in vitro and in vivo, or in chicken and human cells, as mentioned above. Data obtained with NBS1 mutated mice suggest the indirect involvement of NBS1 in V(D)J recombination. In NBS1 mutated mice, when V(D)J recombination occurs at TCR-β and TCR-γ loci, the inter-chromosomal recombination is increased, whereas the intra-chromosomal recombination is not significant affected 21, 22. This is consistent with the frequent chromosomal translocation observed at TCR loci in NBS1 patients 3; suggesting that NBS1 plays a role in recognizing proper elements of V(D)J recombination by suppression of inter-chromosomal recombination, which could lead to gene translocation and genomic instability 3.
In the HR pathway, the NBS1 knockout chicken DT40 cells showed a markedly reduction in HR events (sister chromatid exchange) following mitomycin-C treatment. Moreover, SCneo reporter assays revealed a dramatically reduced HR frequency of approximately 100-fold in NBS1 deficient DT40 cells, as compared to wild-type cells; this suggested that NBS1 is required in HR, at least in vertebrate cells 3, 9, 19.
Roles in cell cycle checkpoint control
NBS1 has proved to be an essential modulator in cell cycle checkpoint control, which is an important part of the DNA damage response. NBS cells exhibit defects in cell cycle checkpoints after exposure to IR or radiomimetic drugs. One of the striking checkpoint defects is in the intra-S-phase checkpoint; the failure of the intra-S-phase checkpoint leads to radio-resistant DNA synthesis (RDS), in which NBS cells continue DNA replication despite the presence of radiation-induced DNA damage 3, 38, 39. There are three parallel pathways to modulate the intra-S-phase checkpoint control 3, 29, 40, 41, 42. The first is ATM-CHK2-CDC25A-CHK2 pathway 40. In this pathway, IR first activates ATM, then ATM activate CHK2, a cell cycle checkpoint kinase, most probably by phosphorylation at thr 68 43, then activated CHK2 phosphorylates CDC25A, another cell cycle signaling kinase at ser 123, which leads to a phosphorylation of CDK2 at tyr 15 and thr 14 and a disruption of cyclin E/CDK2 kinase which causes the intra-S-phase checkpoint 40. NBS1 hase been reported to be required for the activation of ATM and CHK2 after IR 15, 44, 45, 46, 47, 48. The down-regulation of NBS1 by siRNA leads to a decreased activation of CHK2 after exposure to IR 16. Thus, NBS1 could be involved in regulating intra-S-phase checkpoint control via ATM and CHK2. However, the RDS is more severe in AT cells than in NBS cells, and while some reports showed that CHK2 activation was normal in NBS cells 49, others showed that NBS1 was required in CHK2 activation; this suggested that factors other than ATM and CHK2 might also be involved in intra-S-phase checkpoint control.
The second is a pathway involving ATM, NBS1, and a cohesin protein SMC1 51. The effective phosphorylation of SMC1 at ser 957 and ser 966 leads to an activation of an intra-S-phase checkpoint and RDS, which depends on phosphorylation at ser 278 and ser 343 of NBS1 by ATM; this clearly indicated a role of NBS1 in the intra-S-phase checkpoint control 51.
The third is the ATM/FANCD2 pathway 52. In this pathway, ATM phosphorylates ser 222 of FANCD2 (mutated in the chromosomal instability syndrome Fanconi's anemia) and this event activates the intra-S-phase checkpoint. This FANCD2 phosphorylation requires the phosphorylation at ser 343 of NBS1 by ATM 52, 53.
NBS1 may also become involved in G1 checkpoint control although there is some controversy about that. A deficiency in G1 arrest was observed after low doses of IR in primary NBS fibroblasts but was not observed at high doses 54. In addition, the expression of p53 and p21 in NBS fibroblast cell lines, which should lead to G1 cell cycle arrest, was found to be attenuated after IR 55. In B-lymphoblastoid NBS cells, G1 arrest was also found to be defective or attenuated 56, 57. However, some other researchers reported that normal G1 checkpoint control was present in NBS fibroblast cell lines 55, 58. In a mouse model with a mutated NBN (the murine homologue of NBS1), the p21 response in fibroblast cells to IR was found to be indistinguishable from normal control 22.
The reports for NBS cells and the G2 checkpoint are also discrepant. Lymphoblastoid cells derived from NBS patients, unlike normal cells, fail to stop their entry into mitosis immediately after IR and activate CHK2, which suggests a G2 checkpoint defect. The defect of G2 checkpoint and CHK2 activation were rescued by the ectopic expression of a wild-type NBS1 15. NBS lymphoblastoid cells also showed a defect in replication stalling induced G2 checkpoints and the activation of CHK1, c-jun and p53 50. The degradation of MRE11 in Hela cells also leads to a failure of the G2 checkpoint after IR treatment 45. A disruption of G2 checkpoints in mouse fibroblastoid cells with a mutation in NBN was also reported 59 while the EBV transformed B-lymphoblastoid cells from NBS patients the G2 checkpoint was found to be normal 41, 60. Taken together, even though there is some evidence to show that NBS1, and/or the MRN complex is involved in the control of G1 and G2 checkpoints, there is some discrepancy among studies. A possible explanation for this discrepancy is the redundant pathways in checkpoint control, the different cell types tested, and the possible differences among NBS1 mutations used in those studies.
Roles in telomere stability maintenance
Telomeres are highly specialized nucleoprotein structures that maintain genomic stability by stabilizing and protecting the ends of linear chromosomes. Telomeric DNA is composed of tandem arrays of a 5-26 bp repetitive G-rich sequences that are oriented 5′-to-3′ towards the chromosome end; these also serve as binding sites for specific telomere associated proteins 61, 62, 63. Because conventional replication machinery cannot synthesize new DNA to the very end of a linear chromosome, replication results in progressive erosion of telomeric DNA64, 65. Activation of telomerase, a cellular ribonucleoprotein reverse transcriptase responsible for elongating one strand of the telomere, prevents the gradual loss of sequence from chromosome ends 61, 66, 67.
Telomeres “cap” chromosome ends, preventing them from being processed in the same way as broken DNA ends. DSBs normally lead to arrested growth and attempts by the cell to repair the ends 68, 69. Functional telomeres turn this response into an appropriate response that acts to retain genomic integrity 61. Therefore, it is not surprise that DNA DSB repair proteins may play an important role in telomere capping. In normal cells, the background level of DSBs are very low, about 0.05 DSBs/cell 70. However, DNA repair associated proteins are found to participate in capping 92 new telomere ends, which is equivalent to 46 DSBs, far in excess of the background number of DSBs 64. This suggests that DNA repair proteins are involved not only in joining the broken DSBs, but also in preventing the inappropriate joining of telomere ends. A set of proteins which mainly involved in the NHEJ pathway have been demonstrated to prevent telomere dysfunction; these include DNA-PKcs 64, 71, 72, 73, 74, 75, 76, 77, Ku70 73, 75, 76, 77, Ku80 73, 78, 79, 80, XRCC4 80, and Artemis 81, 82.
Accumulating evidence demonstrates that the MRN complex is required for the maintenance of telomere length in mammals, plants and yeast 29, 31, 83, 84, 85, 86, 87. In yeast, the XRS2 complex is involved in at least two pathways in telomere elongation modulation 3, 29, 88, 89. First, it is thought to generate 3′ ssDNA at the telomere, which is essential for the recruitment and subsequent action of telomerase. Second, it is involved in the homologous recombination mediated pathway to elongate telomeres, which involves recombination between tracts of telomere repeats but is not dependent on the function of telomerase 90. In primary fibroblast cells isolated from NBS patients, accelerated telomere shortening during in vitro culture has been observed. Neither the introduction of NBS1 nor the catalytic subunit of telomerase, TERT, alone, has restorative effect on telomere length. Whereas, the co-expression of NBS1 and TERT leads to a significantly greater telomere length, implying that MRN functions in generating the 3′-overhangs and TERT can then replicate the telomeres by using this G-rich strand as a primer 42, 87. However, in T and B cell lines derived from different NBS patients, the telomere length maintenance was found to be intact 91. Due to the fact that a 70 kDa NBS1 variant which contained the MRE11 binding site was present in those patients, the MRE11-binding domain could be sufficient for the telomere length maintenance by NBS1 91. Because NBS is caused by a polymorphism of NBS1 gene, it is reasonable to speculate that different polymorphisms could result in the presence of different NBS1 protein fragments in NBS patients. Thus, the specificity of telomere abnormality could be related to the specificity of individual NBS1 fragments.
In telomerase negative cancer cells, telomere length is maintained by an alternative lengthening of telomeres (ALT). The telomere associated nuclear body, which is dependent on the promyelocytic leukemia protein (PML), is often found in the nucleus of ALT cells. The PML nuclear bodies appear at exactly the same time as the activation of the ALT mechanism during cell immortalization and contain HR associated proteins, such as RPA, RAD51, and RAD52, which suggests that PML may function through a recombination mechanism in ALT 42. NBS1 was found to be co-localized with PML, as well as to be associated with a nuclear dot-associated PML-binding protein, SP100, by the BRCT-domain at its C-terminus 92. NBS1 also functions in recruiting other recombination proteins, including RAD50, MRE11, and BRCA1 to PML nuclear bodies 59, 93. The above mentioned observations suggest a role of NBS1 in the ALT pathway.
In human cells, NBS1 was found to associate with TRF2, an important telomere function modulator, in a cell cycle specific manner 94. Indirect immunofluorescence demonstrated that RAD50 and MRE11 presented at interphase telomeres, whereas NBS1 was associated with TRF2 and telomeres in S-phase, but not in G1 and G2. In mammalian cells, the telomeres end in a large duplex loop (t-loop). Closure and stabilization of the t-loop is thought to occur by the invasion of the telomeric 3′-overhang into the duplex telomeric repeat tract, creating a stable heteroduplex at the base of the loop; that can be viewed as a recombinational event. TRF2 plays an important role in the process of strand invasion and T-loop stabilization. The formation of the telomeric t-loop is required after telomere replication, and the stabilization of the telomeric t-loop is continuously required throughout the cell cycle. Considering the role of MRE11 and RAD50 in HR, it is not surprising to see that MRE11 and RAD50 become involved in this process by functioning in forming and stabilizing telomeric t-loop throughout the whole cell cycle 94. However, the transient recruitment of NBS1 to telomeres only in S-phase suggests a role of NBS1 in telomere replication. NBS1 functions in potentiating DNA-helicase and endonuclease activity of MRE11. Thus, the recruitment of NBS1 to telomeric MRE11/RAD50 may regulate a helicase mediated unpairing of the t-loop base. This process would open the t-loop, perhaps facilitating progression of DNA replication machinery to the end of chromosome 94.
Our recent results showed that the down regulation of NBS1 protein level by siRNA transfection also results in an increase of a telomeric abnormality called telomere association, a phenomenon in which telomeres of the same or different chromosomes are observed in unusually close proximity in metaphase spreads 16. The mechanism of the increased telomere association by NBS1 deficiency is not fully understood. It may related with the functional roles of NBS1/MRN in ALT mediated telomere elongation modulation 42, 59, 92, 93 and the inter-chromosomal recombination of the repetitive telomeric tracts 16.
Modulation of PIKK family
PI3K-like protein kinases (PIKKs) are a conserved family of proteins from yeast to human, most of which possess a serine/threonine kinase activity. All of these proteins contain a domain with motifs that are typical of the lipid kinase phosphatidylinositol 3-kinase (PI3K), so they are termed PIKKs 95. PIKKs respond to various stresses by phosphorylating key proteins in the corresponding response pathways; thus, they trigger numerous processes including cell cycle checkpoint control and DNA repair 95, 96, 97, 98, 99, 100, depending on the spectrum of their downstream substrates. In mammalian cells, the PIKKs members DNA-PKcs, ATM, and ATR are known to be involved in DNA damage response. Whereas ATM and DNA-PKcs responds primarily to DSBs, ATR respond to ultraviolet (UV) light, DSBs, and stalled replication forks 95, 96. Recent studies suggest that NBS1 is required for the activation of the DNA damage pathway of PIKKs family members ATM, ATR, and DNA-Pkcs; direct experimental evidence has been obtained for ATM and ATR 44, 45, 46, 47, 48, 50, 101.
The similarity of clinical manifestation and cellular phenotypic characteristics between NBS and AT patients, namely immunodeficiency, radiation sensitivity, chromosomal instability, and cancer predisposition 7, 9, 102, 103 suggests some possible links between NBS1 and ATM. NBS1 was first recognized as a downstream substrate of ATM based on the evidence that ATM phosphorylated NBS1 at ser 278 and 343 in the central region 15, 104, 105. Phosphorylation of NBS1 by ATM could play some roles in major ATM-mediated pathways, the intra-S and G2/M cell cycle checkpoints 105. Furthermore, NBS1 appears to facilitate ATM-mediated phosphorylation of several ATM substrates, such as the checkpoint kinase CHK2, the chromatin remodeling protein SMC, CHK1, and MRE11 15, 51, 54, 106, 107, 108. However, the fact that MRN complex adherence to the sites of DSB is independent of ATM make people question that ATM acts upstream of NBS1. Uziel et al found that after treatment with the radiomimetic chemical neocarzinostatin, ATM activation measured by the phosphorylation at ser1981 was defective in NBS1 compromised cells. Consistent with that, the nuclear retention of ATM (a sign of chromosomal binding) and the activation of ATM substrates, such as CHK2, P53, and HDM2 were also defective or attenuated in cells with an NBS1 deficiency 48. Carson et al utilized the adenoviral proteins E1b55k/E4orf6 to disrupt the MRN complex in human cells, and showed that after treatment with DSB inducing agents, the infected cells had a marked reduction in ATM phosphorylation, ATM-dependent G2/M checkpoint abrogation, and the ATR-mediated DNA damage response deficiency 45. Similar results were obtained by Horejsi et al, who investigated the timing and magnitude of ATM activation by phosphorylation at ser1981 of ATM and ser 966 of the ATM substrate SMC1 in NBS1 proficient and deficient human fibroblast cells 101. After 2 Gy IR, NBS1 proficient cells showed a rapid phosphorylation of ATM at ser1981 and the substrate. However, the activation of ATM was abrogated or attenuated in NBS1 deficient cells. The activation of ATM and radiosensity was rescued via retrovirally mediated reconstitution of wild-type NBS1, but not NBS1 lacking the MRE11 binding domain 101, which support the view that DNA processing activity of MRE11 is also required for ATM activation 47.
The direct evidence of the physical protein to protein interaction between MRN and ATM, as well as the modulation of the kinase activity ATM was obtained by Lee and Paull 46. By using purified MRN and ATM protein expressed in a baculovirus system, they carried out in vitro kinase activity and gel filtration assays. The results demonstrated that MRN stimulates the kinase activity of ATM in vitro toward its substrates p53, CHK2, and histone H2AX. MRN makes multiple contacts with ATM and stimulates ATM activity by facilitating stable binding to the substrates. To further investigate the role of MRN in the activation of ATM signaling in DSBs, Lee and Paull used a similar assay to test ATM binding to DNA fragments by MRN 47. The results show that the binding of MRN to DNA is ATM independent, whereas ATM is associated with DNA only when MRN was associated. Furthermore, the unwinding of DNA ends by MRN is essential for ATM stimulation. These results indicated that ATM activation by DSB through MRN complex occurred by facilitating ATM-DNA binding and the unwinding of the DNA duplex 47. Stiff et al 50 reported that NBS1 also facilitated ATR-dependent phosphorylation. NBS1 deficient cell lines show a similar defect in ATR phosphorylation of CHK1, c-jun and p53 in response to UV irradiation- and hydroxyurea (HU)-induced replication stalling. NBS1 deficient cells also showed an impaired ubiquitination of FANCD2 after HU treatment, which is ATR dependent. Following HU-induced replication arrest, NBS1 and ATR deficient cells showed a similarly impaired G2/M checkpoint arrest and an impaired ability to restart DNA synthesis at stalled replication forks. Moreover, NBS1 deficient cells failed to retain ATR in the nucleus following HU treatment and extraction 50. A recent finding by Falck et al 44 revealed a conserved C-terminal motif in NBS1 that serves to recruit activated ATM to the sites of DNA damage and promote the phosphorylation of ATM substrates, leading to the events of the DNA damage response. Interestingly, the motif shares sequence homology to the C-terminus of Ku80 and ATRIP. Ku80 and ATRIP are the partner proteins which are required to recruit PIKK family member DNA-PKcs and ATR to the sites of DNA damages, respectively 109, 110, 111, 112, 113. This motif is required for Ku80 and ATRIP interactions with ATR and DNA-PKcs and the modulation of their functions. Interestingly, the synthesized NBS1 C-terminal peptide with this motif interacts with DNA-PKcs as measured by a peptide pull down assay, suggesting a common mechanism of the activation of PIKKs, as well as the possible extension of the role of NBS1 in PIKKs activation 44. In other words, NBS1 may also play an important role in modulating other PIKKs not limited to ATM and ATR; DNA-PKcs could be a reasonable candidate for that.
Conclusions
NBS1 is a component of the MRN complex and plays an important role in DNA damage response. It modulates the DNA damage signal sensing by recruiting PIKK protein family members ATM, ATR, and probably DNA-PKcs to the DNA damage sites and activating their functions. It can also recruit MRE11 and RAD50 to the proximity of DSBs by an interaction with gH2AX through the BRCT/FHA domain at its C-terminus; thus, NBS1 functions in DNA DSB repair both in the NHEJ and HR pathways. NBS1 also functions in telomere length maintenance by generating the 3′ overhang which serves as a primer for telomerase dependent telomere elongation. In addition, NBS1 co-localizes with PML and functions in recruiting recombination proteins to PML to modulate ALT pathway. NBS1 is a major player in the control of intra-S-phase checkpoint and there is some evidence that NBS1 is involved in G1 and G2 checkpoints. The roles of NBS1/MRN encompass DNA damage sensor, signal transducer, and effector, which enable cells to maintain DNA integrity and genomic stability.
References
Abraham RT . Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15:2177–96.
Gollin SM . Mechanisms leading to chromosomal instability. Semin Cancer Biol 2005; 15:33–42.
Tauchi H, Matsuura S, Kobayashi J, et al. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene 2002; 21:8967–80.
Saar K, Chrzanowska KH, Stumm M, et al. The gene for the ataxia-telangiectasia variant, Nijmegen breakage syndrome, maps to a 1-cm interval on chromosome 8q21. Am J Hum Genet 1997; 60:605–10.
Varon R, Vissinga C, Platzer M, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998; 93:467–76.
Distel L, Neubauer S, Varon R, et al. Fatal toxicity following radio- and chemotherapy of medulloblastoma in a child with unrecognized Nijmegen breakage syndrome. Med Pediatr Oncol 2003; 41:44–8.
Tauchi H, Matsuura S, Isomura M, et al. Sequence analysis of an 800-kb genomic DNA region on chromosome 8q21 that contains the Nijmegen breakage syndrome gene, NBS1. Genomics 1999; 55:242–7.
Dumon-Jones V, Frappart PO, Tong WM, et al. NBN heterozygosity renders mice susceptible to tumor formation and ionizing radiation-induced tumorigenesis. Cancer Res 2003; 63:7263–9.
Tauchi H, Kobayashi J, Morishima K, et al. NBS1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 2002; 420:93–8.
Taylor AM . Chromosome instability syndromes. Best Pract Res Clin Haematol 2001; 14:631–44.
Weemaes CM, Smeets DF, van der Burgt CJ . Nijmegen breakage syndrome: A progress report. Int J Radiat Biol 1994; 66:S185–8.
Wilda M, Demuth I, Concannon P, et al. Expression pattern of the Nijmegen breakage syndrome gene, NBS1, during murine development. Hum Mol Genet 2000; 9:1739–44.
Seidemann K, Tiemann M, Henze G, et al. Therapy for non-hodgkin lymphoma in children with primary immunodeficiency: Analysis of 19 patients from the NFM trials. Med Pediatr Oncol 1999; 33:536–44.
Resnick IB, Kondratenko I, Togoev O, et al. Nijmegen breakage syndrome: Clinical characteristics and mutation analysis in eight unrelated Russian families. J Pediatr 2002; 140:355–61.
Buscemi G, Savio C, Zannini L, et al. CHK2 activation dependence on NBS1 after DNA damage. Mol Cell Biol 2001; 21:5214–22.
Zhang Y, Lim CUK, Williams ES, et al. NBS1 knockdown by small interfering RNA increases ionizing radiation mutagenesis and telomere association in human cells. Cancer Res 2005; 65:5544–53.
Varon R, Seemanova E, Chrzanowska K, et al. Clinical ascertainment of Nijmegen breakage syndrome (NBS) and prevalence of the major mutation, 657del5, in three Slav populations. Eur J Hum Genet 2000; 8:900–2.
Kobayashi J, Tauchi H, Sakamoto S, et al. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 2002; 12:1846–51.
Matsuura S, Kobayashi J, Tauchi H, Komatsu K . Nijmegen breakage syndrome and DNA double strand break repair by NBS1 complex. Adv Biophys 2004; 38:65–80.
Desai-Mehta A, Cerosaletti KM, Concannon P . Distinct functional domains of Nibrin mediate MRE11 binding, focus formation, and nuclear localization. Mol Cell Biol 2001; 21:2184–91.
Kang J, Bronson RT, Xu Y . Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. Embo J 2002; 21:1447–55.
Williams BR, Mirzoeva OK, Morgan WF, et al. A murine model of Nijmegen breakage syndrome. Curr Biol 2002; 12:648–53.
Maser RS, Zinkel R, Petrini JH . An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat Genet 2001; 27:417–21.
Dasika GK, Lin SC, Zhao S, et al. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene 1999; 18:7883–99.
Ferguson DO, Alt FW . DNA double strand break repair and chromosomal translocation: Lessons from animal models. Oncogene 2001; 20:5572–9.
Collis SJ, DeWeese TL, Jeggo PA, Parker AR . The life and death of DNA-PK. Oncogene 2005; 24:949–61.
Paull TT, Gellert M . The 3′ to 5′ exonuclease activity of MRE11 facilitates repair of DNA double-strand breaks. Mol Cell 1998; 1:969–79.
Paull TT, Gellert M . NBS1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the MRE11/RAD50 complex. Genes Dev 1999; 13:1276–88.
D'Amours D, Jackson SP . The MRE11 complex: At the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002; 3:317–27.
Haber JE . The many interfaces of MRE11. Cell 1998; 95:583–6.
Boulton SJ, Jackson SP . Components of the ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. Embo J 1998; 17:1819–28.
Ueno M, Nakazaki T, Akamatsu Y, et al. Molecular characterization of the schizosaccharomyces pombe NBS1 gene involved in DNA repair and telomere maintenance. Mol Cell Biol 2003; 23:6553–63.
Clatworthy AE, Valencia-Burton MA, Haber JE, Oettinger MA . The MRE11-RAD50-XRS2 complex, in addition to other non-homologous end-joining factors, is required for V(D)J joining in yeast. J Biol Chem 2005; 280:20247–52.
Ohta K, Nicolas A, Furuse M, et al. Mutations in the MRE11, RAD50, XRS2, and MRE2 genes alter chromatin configuration at meiotic DNA double-stranded break sites in premeiotic and meiotic cells. Proc Natl Acad Sci USA 1998; 95:646–51.
Huang J, Dynan WS . Reconstitution of the mammalian DNA double-strand break end-joining reaction reveals a requirement for an MRE11/RAD50/NBS1-containing fraction. Nucleic Acids Res 2002; 30:667–74.
Udayakumar D, Bladen CL, Hudson FZ, Dynan WS . Distinct pathways of nonhomologous end joining that are differentially regulated by DNA-dependent protein kinase-mediated phosphorylation. J Biol Chem 2003; 278:41631–5.
Harfst E, Cooper S, Neubauer S, et al. Normal V(D)J recombination in cells from patients with Nijmegen breakage syndrome. Molecular Immunology 2000; 37:915–29.
Shiloh Y . ATM and related protein kinases: Safeguarding genome integrity. Nat Rev Cancer 2003; 3:155–68.
Shiloh Y . ATM: Ready, set, go. Cell Cycle 2003; 2:116–7.
Weizman N, Shiloh Y, Barzilai A . Contribution of the ATM protein to maintaining cellular homeostasis evidenced by continuous activation of the AP-1 pathway in ATM-deficient brains. J Biol Chem 2003; 278:6741–7.
Abraham RT . PI 3-kinase related kinases: 'big' players in stress-induced signaling pathways. DNA Repair (Amst) 2004; 3:883–7.
Bakkenist CJ, Kastan MB . Initiating cellular stress responses. Cell 2004; 118:9–17.
Goodarzi AA, Block WD, Lees-Miller SP . The role of ATM and ATR in DNA damage-induced cell cycle control. Prog Cell Cycle Res 2003; 5:393–411.
Falck J, Coates J, Jackson SP . Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005; 434:605–11.
Uziel T, Lerenthal Y, Moyal L, et al. Requirement of the MRN complex for ATM activation by DNA damage. Embo J 2003; 22:5612–21.
Carson CT, Schwartz RA, Stracker TH, et al. The MRE11 complex is required for ATM activation and the G2/M checkpoint. Embo J 2003; 22:6610–20.
Lee JH, Paull TT . ATM activation by DNA double-strand breaks through the MRE11-RAD50-NBS1 complex. Science 2005; 308:551–4.
Lee JH, Paull TT . Direct activation of the ATM protein kinase by the MRE11/RAD50/NBS1 complex. Science 2004; 304:93–6.
Horejsi Z, Falck J, Bakkenist CJ, et al. Distinct functional domains of NBS1 modulate the timing and magnitude of ATM activation after low doses of ionizing radiation. Oncogene 2004; 23:3122–7.
Stiff T, Reis C, Alderton GK, et al. NBS1 is required for ATR-dependent phosphorylation events. Embo J 2005; 24:199–208.
Tauchi H, Kobayashi J, Morishima K, et al. The forkhead-associated domain of NBS1 is essential for nuclear foci formation after irradiation but not essential for hRAD50/hMRE11/NBS1 complex DNA repair activity. J Biol Chem 2001; 276:12–5.
Lavin MF, Birrell G, Chen P, et al. ATM signaling and genomic stability in response to DNA damage. Mutat Res 2005; 569:123–32.
Lim DS, Kim ST, Xu B, et al. ATM phosphorylates p95/NBS1 in an S-phase checkpoint pathway. Nature 2000; 404:613–7.
Zhao S, Weng YC, Yuan SS, et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature 2000; 405:473–7.
Girard PM, Riballo E, Begg AC, et al. NBS1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene 2002; 21:4191–9.
Lee JH, Xu B, Lee CH, et al. Distinct functions of Nijmegen breakage syndrome in ataxia telangiectasia mutated-dependent responses to DNA damage. Mol Cancer Res 2003; 1:674–81.
Yazdi PT, Wang Y, Zhao S, et al. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev 2002; 16:571–82.
Gatei M, Sloper K, Sorensen C, et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of CHK1 on ser-317 in response to ionizing radiation. J Biol Chem 2003; 278:14806–11.
Dong Z, Zhong Q, Chen PL . The Nijmegen breakage syndrome protein is essential for MRE11 phosphorylation upon DNA damage. J Biol Chem 1999; 274:19513–6.
Unsal-Kacmaz K, Sancar A . Quaternary structure of ATR and effects of ATRIP and replication protein a on its DNA binding and kinase activities. Mol Cell Biol 2004; 24:1292–300.
Osborn AJ, Elledge SJ, Zou L . Checking on the fork: The DNA-replication stress-response pathway. Trends Cell Biol 2002; 12:509–16.
Zou L, Elledge SJ . Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542–8.
Gottlieb TM, Jackson SP . The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993; 72:131–42.
Dvir A, Peterson SR, Knuth MW, et al. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase ii. Proc Natl Acad Sci USA 1992; 89:11920–4.
McEachern MJ, Krauskopf A, Blackburn EH . Telomeres and their control. Annu Rev Genet 2000; 34:331–58.
Blackburn EH . Switching and signaling at the telomere. Cell 2001; 106:661–73.
Bai Y, Murnane JP . Telomere instability in a human tumor cell line expressing NBS1 with mutations at sites phosphorylated by atm. Mol Cancer Res 2003; 1:1058–69.
Bailey SM, Goodwin EH . DNA and telomeres: Beginnings and endings. Cytogenet Genome Res 2004; 104:109–15.
Watson JD . Origin of concatemeric t7 DNA. Nat New Biol 1972; 239:197–201.
Greider CW, Blackburn EH . Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 1985; 43:405–13.
Yu GL, Bradley JD, Attardi LD, Blackburn EH . In vivo alteration of telomere sequences and senescence caused by mutated tetrahymena telomerase RNAs. Nature 1990; 344:126–32.
Bennett CB, Lewis AL, Baldwin KK, Resnick MA . Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc Natl Acad Sci USA 1993; 90:5613–7.
Sandell LL, Zakian VA . Loss of a yeast telomere: Arrest, recovery, and chromosome loss. Cell 1993; 75:729–39.
Rothkamm K, Lobrich M . Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA 2003; 100:5057–62.
Bailey SM, Brenneman MA, Halbrook J, et al. The kinase activity of DNA-PK is required to protect mammalian telomeres. DNA Repair (Amst) 2004; 3:225–33.
Bailey SM, Cornforth MN, Ullrich RL, Goodwin EH . Dysfunctional mammalian telomeres join with DNA double-strand breaks. DNA Repair (Amst) 2004; 3:349–57.
Bailey SM, Meyne J, Chen DJ, et al. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc Natl Acad Sci USA 1999; 96:14899–904.
Lees-Miller SP, Godbout R, Chan DW, et al. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science 1995; 267:1183–5.
Gilley D, Tanaka H, Hande MP, et al. DNA-PKcs is critical for telomere capping. Proc Natl Acad Sci USA 2001; 98:15084–8.
Hsu HL, Gilley D, Galande SA, et al. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev 2000; 14:2807–12.
Hsu HL, Gilley D, Blackburn EH, Chen DJ . Ku is associated with the telomere in mammals. Proc Natl Acad Sci USA 1999; 96:12454–8.
Samper E, Goytisolo FA, Slijepcevic P, et al. Mammalian ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep 2000; 1:244–52.
Espejel S, Franco S, Rodriguez-Perales S, et al. Mammalian ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. Embo J 2002; 21:2207–19.
d'Adda di Fagagna F, Hande MP, Tong WM, et al. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr Biol 2001; 11:1192–6.
Rooney S, Sekiguchi J, Zhu C, et al. Leaky SCID phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol Cell 2002; 10:1379–90.
Rooney S, Alt FW, Lombard D, et al. Defective DNA repair and increased genomic instability in Artemis-deficient murine cells. J Exp Med 2003; 197:553–65.
Le S, Moore JK, Haber JE, Greider CW . RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 1999; 152:143–52.
Kironmai KM, Muniyappa K . Alteration of telomeric sequences and senescence caused by mutations in RAD50 of saccharomyces cerevisiae. Genes Cells 1997; 2:443–55.
Nugent CI, Bosco G, Ross LO, et al. Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr Biol 1998; 8:657–60.
Gallego ME, White CI . RAD50 function is essential for telomere maintenance in Arabidopsis. Proc Natl Acad Sci USA 2001; 98:1711–6.
Ranganathan V, Heine WF, Ciccone DN, et al. Rescue of a telomere length defect of Nijmegen breakage syndrome cells requires NBS and telomerase catalytic subunit. Curr Biol 2001; 11:962–6.
de Lange T . Protection of mammalian telomeres. Oncogene 2002; 21:532–40.
de Lange T . T-loops and the origin of telomeres. Nat Rev Mol Cell Biol 2004; 5:323–9.
Kass-Eisler A, Greider CW . Recombination in telomere-length maintenance. Trends Biochem Sci 2000; 25:200–4.
Iijima K, Komatsu K, Matsuura S, Tauchi H . The Nijmegen breakage syndrome gene and its role in genome stability. Chromosoma 2004; 113:53–61.
Siwicki JK, Degerman S, Chrzanowska KH, Roos G . Telomere maintenance and cell cycle regulation in spontaneously immortalized T-cell lines from Nijmegen breakage syndrome patients. Exp Cell Res 2003; 287:178–89.
Naka K, Ikeda K, Motoyama N . Recruitment of NBS1 into PML oncogenic domains via interaction with sp100 protein. Biochem Biophys Res Commun 2002; 299:863–71.
Wu G, Lee WH, Chen PL . NBS1 and TRF1 colocalize at promyelocytic leukemia bodies during late S/G2 phases in immortalized telomerase-negative cells. Implication of NBS1 in alternative lengthening of telomeres. J Biol Chem 2000; 275:30618–22.
Wu X, Ranganathan V, Weisman DS, et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature 2000; 405:477–82.
Zhu XD, Kuster B, Mann M, et al. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet 2000; 25:347–52.
Lavin MF, Schroeder AL . Damage-resistant DNA synthesis in eukaryotes. Mutat Res 1988; 193:193–206.
Painter RB, Young BR . Radiosensitivity in ataxia-telangiectasia: A new explanation. Proc Natl Acad Sci USA 1980; 77:7315–7.
Falck J, Mailand N, Syljuasen RG, et al. The ATM-CHK2-CDC25a checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410:842–7.
Digweed M, Sperling K . Nijmegen breakage syndrome: Clinical manifestation of defective response to DNA double-strand breaks. DNA Repair (Amst) 2004; 3:1207–17.
Matsuoka S, Rotman G, Ogawa A, et al. Ataxia telangiectasia-mutated phosphorylates CHK2 in vivo and in vitro. Proc Natl Acad Sci USA 2000; 97:10389–94.
Falck J, Petrini JH, Williams BR, et al. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet 2002; 30:290–4.
Kobayashi J, Antoccia A, Tauchi H, et al. NBS1 and its functional role in the DNA damage response. DNA Repair (Amst) 2004; 3:855–61.
Nakanishi K, Taniguchi T, Ranganathan V, et al. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol 2002; 4:913–20.
Little JB, Nagasawa H, Dahlberg WK, et al. Differing responses of Nijmegen breakage syndrome and ataxia telangiectasia cells to ionizing radiation. Radiat Res 2002; 158:319–26.
Antoccia A, Stumm M, Saar K, et al. Impaired p53-mediated DNA damage response, cell-cycle disturbance and chromosome aberrations in Nijmegen breakage syndrome lymphoblastoid cell lines. Int J Radiat Biol 1999; 75:583–91.
Sullivan KE, Veksler E, Lederman H, Lees-Miller SP . Cell cycle checkpoints and DNA repair in Nijmegen breakage syndrome. Clin Immunol Immunopathol 1997; 82:43–8.
Yamazaki V, Wegner RD, Kirchgessner CU . Characterization of cell cycle checkpoint responses after ionizing radiation in Nijmegen breakage syndrome cells. Cancer Res 1998; 58:2316–22.
Xu B, Kim S, Kastan MB . Involvement of BRCA1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol 2001; 21:3445–50.
Chen L, Trujillo K, Ramos W, et al. Promotion of DNl4-catalyzed DNA end-joining by the RAD50/MRE11/XRS2 and HDF11/HDF2 complexes. Mol Cell 2001; 8:1105–15.
Acknowledgements
We thank Dr Howard L Liber and Dr Susan M Bailey for their helpful suggestions and comments during the preparation of this manuscript. We apologize to many authors of the outstanding papers that were not cited here due to space limitation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, Y., Zhou, J. & Lim, C. The role of NBS1 in DNA double strand break repair, telomere stability, and cell cycle checkpoint control. Cell Res 16, 45–54 (2006). https://doi.org/10.1038/sj.cr.7310007
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310007
Keywords
This article is cited by
-
TRIM24 is critical for the cellular response to DNA double-strand breaks through regulating the recruitment of MRN complex
Oncogene (2023)
-
The role of forkhead-associated (FHA)-domain proteins in plant biology
Plant Molecular Biology (2023)
-
Validation of the γH2AX biomarker for genotoxicity assessment: a review
Archives of Toxicology (2019)
-
DNA single-strand break-induced DNA damage response causes heart failure
Nature Communications (2017)
-
Genomic integrity and the ageing brain
Nature Reviews Neuroscience (2015)
