
Abstract
Genetic diversity at 10 loci encoding six enzymes was studied in 17 European populations of Sorbus aucuparia L. (the rowan), distributed among five regions, from Finland to the Pyrenees. Levels of genetic diversity were high both at the species level (He=0.229) and within populations (mean He=0.212), whereas levels of differentiation among populations were very low (GST=0.060). These values were comparable to those observed in other woody species with similar ecological traits. Populations from the Plateau des Tailles (Belgium) exhibited an unexpectedly high level of differentiation among populations, which was tentatively explained by the recent history of colonization experienced by these populations. Despite the low overall differentiation, geographical patterns were observed. Gene flow estimates and geographical distances between pairs of populations appeared to be correlated (r=−0.483), resulting in a pattern of isolation by distance, which was disrupted to some extent by the local dynamics of the Tailles populations. Cluster analysis revealed that populations were genetically more similar to populations from the same region compared to populations from other geographical groups. Finnish populations, in particular, appeared to be highly differentiated from more southerly populations.
Similar content being viewed by others

Introduction
Over the past two decades, a growing number of investigations on the patterns of genetic variation within tree species have been performed. Hence, the availability of considerable data based on a common methodology, i.e. isozyme electrophoresis, has motivated the search for general patterns of population genetic structure among woody perennial species (e.g. Hamrick & Godt, 1989; Hamrick et al., 1992). Compared to nonwoody species, woody perennials maintain significantly more variation within species and populations, but show lower differentiation among populations. Despite average levels of genetic differentiation among populations as low as 10% of the total variation, geographical patterns of variation have often been detected in studies covering a wide geographical scale (Lagercrantz & Ryman, 1990; Comps et al., 1991; Zanetto & Kremer, 1995).
The review by Hamrick et al. (1992) on the factors influencing levels of genetic diversity in woody species stressed the importance of geographical range and, to a lesser extent, of mating system and seed dispersal mechanism. Only 16% of the variation in the level of differentiation among populations (measured by GST) was explained by the seven biological traits investigated by Hamrick et al. (1992), which suggests that other factors play a significant role in determining how genetic variation is distributed within and among populations. Among these factors, the evolutionary history of species must have a major influence (Hamrick et al., 1992). According to Bergmann (1991), the most important aspects of species evolutionary history in shaping the patterns of genetic variation within European forest tree species are: (i) the number and types of glacial refugia; (ii) the migration routes during postglacial periods; and (iii) human activities (silvicultural practices in particular). Range-wide patterns of genetic diversity have, however, been studied in only a few European tree species, which consist almost exclusively of those of economic importance such as beech (Comps et al., 1991), oaks (Zanetto & Kremer, 1995) and Norway spruce (Lagercrantz & Ryman, 1990).
Sorbus aucuparia (rowan) is a small, insect-pollinated, self-incompatible tree (Raspé, 1998) widespread in Europe, from Iceland and northern Russia to the mountains of central Spain, Portugal, Italy and the Caucasus, as well as in northern Asia Minor (Clapham et al., 1962). The species is at its best in a relatively wet (rainfall min. 750 mm yr−1), cool climate (Rameau et al., 1989). Consequently, it is confined to mountainous areas in the most southern part of its range. The seeds are dispersed mainly by birds (Snow & Snow, 1988), but also by mammals (Grime et al., 1988). In many places (mainly at lower altitudes) S. aucuparia often behaves as a hardy pioneer or postpioneer species (Kullman, 1986; Rameau et al., 1989), populations of which are later replaced by late successional tree populations. At higher altitudes, however, it is one of the few species which can maintain the tree habit and its populations may be part of the late successional vegetation. The ecology of S. aucuparia, therefore, contrasts to some extent with the previously studied tree species cited above, which are all wind-pollinated, late successional species.
The purpose of this study is to quantify the genetic variation within and among populations of S. aucuparia using allozyme markers. The sampling design, which consisted of several populations within each of five regions distributed over a large geographical scale, was aimed at detecting potential heterogeneity among groups of populations in the level and spatial pattern of genetic variation.
Materials and methods
Plant material
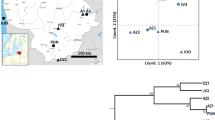
A total of 17 populations were sampled from five regions in Europe: three populations from the Pyrenees; three populations from Auvergne (Central France); two populations from Alsace (East France); four populations from the Plateau des Tailles (East Belgium); and five populations from Finland (Fig. 1). From 25 to 51 individuals were sampled per population, a total of 645 individuals. The sampled populations were representative of a wide range of ecological conditions (Table 1). Geographical distances between populations within regions ranged from 1.8 to 536 km.

Geographical distribution of sampled populations of Sorbus aucuparia. Py, Pyrenees; Au, Auvergne; Al, Alsace; Ta, Plateau des Tailles; Fi, Finland.
Electrophoresis procedures
Tissue from breaking buds was analysed by starch gel electrophoresis according to Raspé et al. (1998). Genotypes were obtained for 10 loci coding for six enzymes: alcohol dehydrogenase (ADH, EC 1.1.1.1: Adh-1), aspartate aminotransferase (AAT, EC 2.6.1.1: Aat-1), isocitrate dehydrogenase (IDH, EC 1.1.1.42: Idh-1, Idh-2), peroxidase (PRX, EC 1.11.1.7: Prx-1), phosphoglucomutase (PGM, EC 5.4.2.2: Pgm-1, Pgm-2, Pgm-3) and 6-phosphogluconate dehydrogenase (6PGD, EC 1.1.1.44: 6Pgd-1, 6Pgd-2). Genetic analysis of eight of these loci (Aat-1, Adh-1, Idh-1, Prx-1, 6Pgd-2, Pgm-1, Pgm-2, Pgm-3) was in accordance with a single-locus codominant mode of inheritance, except for Pgm-2, in which partial dominance (because of a recessive null allele) had to be included in the mode of inheritance (Raspé et al., 1998). However, it was decided to include this locus in the analysis because it showed no peculiarities for parameters that could have been affected by the presence of a null allele. More specifically, the inbreeding coefficient was low and not significantly different from zero in all populations and therefore it could reasonably be assumed that the frequency of the null allele in populations other than the one tested in Raspé et al. (1998) should be very low, if not zero.
Data analysis
Except when otherwise stated, all computations and statistical tests were performed using GEN-SURVEY, a program written by X. Vekemans (Vekemans & Lefèbvre, 1997). Classical estimates of genetic variation were computed within each of the 17 populations, as well as at the species level: percentage of loci polymorphic at the 95% level (P), mean number of alleles per locus (A), and expected heterozygosity (He), which was corrected for small sample size (Nei, 1978). For each locus in every population, the inbreeding coefficient (FIS) was computed following Kirby (1975), and an exact test for departure from Hardy–Weinberg equilibrium was performed using GENEPOP 3 (Raymond & Rousset, 1995a). The rejection level was adjusted for multiple tests by the Dunn-Sˇidák method (Sokal & Rohlf, 1995), as follows:

where k is the number of tests.
Genetic differentiation among all populations, as well as among populations within regions, was investigated with two approaches. An exact test for homogeneity of allele frequencies among populations was performed using GENEPOP 3 (Raymond & Rousset, 1995a). Gene diversity analysis after Nei (1973) with corrections for small sample size (Nei & Chesser, 1983) was also performed. HT, HS and GST were computed for each locus and confidence intervals at 95% were obtained for means over loci by bootstrapping over loci. Furthermore, hierarchical gene diversity analysis was performed by partitioning genetic variation (HT) into three components: within populations (HS), among populations within regions (DSR), and among regions (DRT), with DSR+ DRT=DST. Idh-2 was excluded from all gene diversity analyses, because only two copies of the gene (out of 1290) were found to be variable. Past gene flow, expressed as number of migrants per generation (Nm), was estimated by the private allele method (Slatkin, 1985) using the formula:
ln p(1)=a ln (Nm)+b,
where p(1) is the mean frequency of private alleles (i.e. alleles restricted to a single population), and a and b are coefficients which depend on the number of individuals sampled from each population. The coefficients a and b were estimated according to Barton & Slatkin (1986) using GENEPOP 3 (Raymond & Rousset, 1995a).
The pattern of gene flow among populations was studied according to the method of Slatkin (1993) based on the model of isolation by distance. The extent of gene flow between two populations was estimated as M=[(1/FST) −1]/4, where FST was estimated according to Weir & Cockerham (1984). The significance of the correlation between a similarity matrix with pairwise M-values and a dissimilarity matrix with geographical distances was tested by a Mantel test. The Mantel statistic Z was standardized into a product-moment correlation coefficient (r). A simple regression analysis of log10 (M) was also performed on the logarithm of geographical distance (Slatkin, 1993). To describe further the geographical pattern of genetic variation, BIOSYS-1 (Swofford & Selander, 1981) was used to perform a UPGMA cluster analysis on the Cavalli-Sforza & Edwards (1967) arc distances between population pairs.
Genetic differentiation between the group of Finnish populations and a group including the other, more southerly, populations was tested a posteriori (see below) as follows (Vekemans & Lefèbvre, 1997). Genetic distances between pairs of populations were computed as the coancestry coefficient (Reynolds et al., 1983), which were then averaged separately for pairwise comparisons within and between each group. Statistical significance of the difference between the average-distance-within against average-distance-between groups was investigated using a permutation method (Sokal & Rohlf, 1995): one thousand samples were simulated by randomly assigning populations to each group in order to build the distribution of the statistic under interest, then the observed difference was tested against two-tailed critical values at 95% and 99% of the distribution.
Results
Allozyme diversity at population and species levels
A total of 37 alleles were scored at the 10 loci across the 17 populations analysed. The number of alleles detected at each locus ranged from two (Idh-2 and Pgm-3) to six (6Pgd-2), with a mean of 3.7 (SD=1.25). The table of allelic frequencies is available from the authors on request. Levels of genetic variability within each population are detailed in Table 2. The ranges of percentage of polymorphic loci (P), mean number of alleles per locus (A) and expected heterozygosity (He) were 50–80%, 1.9–2.6 and 0.159–0.277, respectively. Although the coefficient of variation (CV) among populations reached 15.7%, 16% and 6.7% for P, He and A, respectively, no significant differences were generally observed among regions. An apparently significant exception to this homogeneity was the lower level of expected heterozygosity within Finnish populations compared to more southerly populations. This difference, however, appeared to result mainly from higher frequencies of the common allele at only two loci (Pgm-2 and Idh-1), which were highly polymorphic in the other populations. Values of P, A and He were 62.9%, 2.25 and 0.212, respectively, when averaged over populations and 90%, 3.7 and 0.229 at the species level (Table 2). The inbreeding coefficient (FIS) ranged from −0.132 in Ta-2 (Belgium) to 0.069 in Au-3 (Auvergne, France), with a mean of −0.007 over all populations. The ratio of positive to negative coefficients was 8: 9, i.e. as close as possible to a 1: 1 ratio. Out of the 124 exact tests for departure from Hardy–Weinberg expectations, only one was significant (P=0.0003) after adjustment of the rejection level (α§=0.0004 for α=0.05). This test concerned Idh-1 in Ta-1 and corresponded to a FIS of −0.047. These analyses indicate that there is no tendency towards heterozygote excess or deficiency within populations of S. aucuparia.
Spatial structure of variation and gene flow
Values of GST among the 17 populations were consistently low over the 10 loci analysed, with an average of 0.060 (Table 3). However, exact tests for homogeneity of allele frequencies among populations revealed highly significant heterogeneity for all loci (Table 3). Most of the variation among populations (DST) was distributed among regions (86%), with only little (14%) among populations within regions (DSR=0.0024, DRT=0.0149). Some heterogeneity in the level of genetic differentiation among populations within a region was observed among regions (Table 4). Interestingly, the Tailles populations exhibited the highest level of differentiation, although the mean geographical distance between them was lower than in any other region (Table 4). However, the differentiation within the Tailles region was significantly higher (nonoverlapping confidence intervals) only when compared to Auvergne. This was confirmed by tests of homogeneity of allele frequencies (highly significant heterogeneity for the Pyrenees, Tailles and Finland but nonsignificant for Auvergne and Alsace; Table 4).
Gene flow among the 17 populations, estimated using Slatkin's private allele method, was high (Nm=4.62). A significant correlation was observed between matrices of pairwise geographical distances and gene flow estimates between pairs of populations (Mantel test; r=−0.483, P<0.0001). The simple regression analysis on log-transformed data yielded the following relationship between gene flow (M) and geographical distance (D): log10 (M)=−0.259 log10 (D)+1.333, with the model explaining 23% of the variation (R2=0.233). These results indicate a moderate but significant pattern of isolation by distance.
As shown above, the Tailles populations exhibited an unexpectedly high level of differentiation (and consequently a low level of gene flow) given the small distances between these populations. Therefore tests were undertaken for isolation by distance as above but with the Tailles populations excluded from the analysis. This exclusion resulted in an apparently much greater isolation by distance (r = −0.666; log10 (M)=−0.467 log10 (D)+2.019, R2=0.444).
In the dendrogram obtained by UPGMA clustering based on Cavalli-Sforza & Edwards (1967) arc distances, populations tended to cluster according to geographical regions with two exceptions: Py-3 and Ta-3, which clustered with populations from Auvergne. Moreover, Finnish populations were separated out at the first level (Fig. 2). The mean pairwise genetic distance between these two groups (Finnish vs. other populations) was significantly greater (P<0.0001) than the mean pairwise genetic distance within groups, which confirms that the Finnish populations are genetically differentiated from the more southerly populations.

UPGMA cluster analysis of Cavalli-Sforza & Edwards (1967) arc distances for the 17 populations of Sorbus aucuparia.
Discussion
Allozyme diversity at population and species levels
As pointed out by Hamrick & Godt (1989) and Hamrick et al. (1992), long-lived woody perennials usually maintain higher levels of genetic variation within populations, as well as at the species level, compared to species with other life forms. In comparison to the mean values given by Hamrick et al. (1992) for long-lived woody perennials (P=49.3%, A=1.76, He=0.148 within populations; P=65.0%, A=2.22, He=0.177 within species), estimates of genetic variation were higher in S. aucuparia for all parameters, both within populations (P=62.9%, A=2.25, He=0.212) and within species (P=90%, A=3.70, He=0.229). Among woody plants, those species with large geographical ranges, outcrossing breeding system and seed dispersal via animal-ingestion, maintain more genetic diversity within species and populations than woody species with other combinations of traits (Hamrick et al., 1992). Sorbus aucuparia shows this particular combination of traits. The high level of genetic variation observed in the present study seems, therefore, consistent with the ecological attributes of the species, and does not suggest the occurrence of strong genetic bottlenecks during postglacial recolonization. Although Finnish populations appeared to show lower heterozygosity within populations than southerly populations, the overall heterogeneity among populations in levels of He (CV=16%) was typical for an outcrossing plant species (CV=12±3%) as compared to the high heterogeneity observed within selfing species (CV=64±15%) (Schoen & Brown, 1991).
Spatial structure of variation and gene flow
The overall level of differentiation, as measured by GST, was very low; only 6.0% of the total diversity was attributable to interpopulation variation. This value is similar to the mean for woody species, especially when compared to species sharing the same ecological and life history traits (Hamrick et al., 1992) and suggests high levels of gene flow in S. aucuparia. High levels of gene flow through seed and pollen dispersal are likely in this species, because seeds can be dispersed by birds and mammals which feed on the fleshy fruits (Grime et al., 1988; Snow & Snow, 1988). Moreover, S. aucuparia is insect-pollinated and self-incompatible (Raspé, 1998) and, although insect pollination was previously thought to result in very restricted pollen dispersal (e.g. Levin & Kerster, 1974), a recent review of direct estimates of pollen flow using genetic markers (Hamrick et al., 1995) suggested that gene flow mediated by insect pollination could be extensive among populations separated by more than 1000 m. It should be remembered, however, that levels of gene flow inferred from genetic structure of populations (as in this study) actually reflect past gene flow and integrate both realized gene flow through seed and pollen dispersal between established populations and the evolutionary history of the populations (Slatkin, 1987). Furthermore, the influence of the evolutionary history of populations is probably stronger for long-lived (e.g. trees) than for short-lived species, because fewer generations are likely to have followed each other after major historical events such as postglacial recolonization. In Finland, S. aucuparia shows an almost continuous distribution (P. Vakkari, pers. comm.), but in southern France it is confined to mountainous areas because of its climatic requirements. Consequently, important discontinuities arise in the distribution of the species among the southern regions (the Pyrenees, Auvergne, and to a lesser extent Alsace). Therefore, the low level of genetic differentiation observed over the whole sample might reflect contemporary gene flow among Finnish populations, whereas in southern areas it might represent both present-day gene flow among populations within regions and historical gene flow that occurred among regions at a time when the climate was cooler and wetter in southern Europe, allowing a more continuous distribution of the species. Finally, because S. aucuparia is often planted for ornamental purposes, man-mediated gene flow cannot be excluded a priori and might, in part, be responsible for the low levels of differentiation observed.
It has been shown that some heterogeneity in the level of differentiation among populations was apparent among regions. In particular, differentiation among the Tailles populations was unexpectedly high given the high proximity of these populations. It is interesting to note that the populations sampled from the Plateau des Tailles have originated from the recent colonization of clear-fellings in Picea abies plantations (probably less than 25 years ago). It may therefore be asked whether the relatively high differentiation could result from these recent colonization events. Indeed, theoretical studies have shown that founding events may increase differentiation among young populations, depending notably on the number of individuals involved in the typical founding event and on the number of source populations from which they are drawn (Wade & McCauley, 1988; Whitlock & McCauley, 1990). A few studies have tested the predictions of Wade and McCauley on metapopulations of weedy species and reported that colonization dynamics indeed increased genetic variance between populations (McCauley et al., 1995; Giles & Goudet, 1997).
Isolation by distance and geographical patterns
Although only weak differentiation was found among the 17 populations analysed, some geographical patterns of variation could be observed. First, when relating gene flow between populations and geographical distances, a pattern of isolation by distance was detected. This does not necessarily mean that the populations are at equilibrium between drift and migration, because other processes can lead to apparent isolation by distance (Slatkin, 1993). Without direct observations of dispersal, it cannot be certain that the estimates of gene flow from the present study actually reflect effective dispersal. Moreover, local processes occurring at the Plateau des Tailles which increased genetic differentiation among populations may obviously have disrupted the pattern of isolation by distance.
Secondly, the present study showed by use of cluster analysis that populations were more similar genetically to populations from the same region compared to populations from other geographical groups. Finnish populations, in particular, appeared to be highly significantly differentiated from more southerly populations. Two hypotheses can be proposed to account for the divergence of Finnish populations. On the one hand, the greater geographical distance separating the latter from the southerly populations might be sufficient to allow for the observed divergence to occur under isolation by distance. On the other hand, the two groups of populations might have originated from different glacial refugia (or postglacial colonization pathways). The importance of the latter hypothesis has been emphasized in several studies of other tree species (e.g. Comps et al., 1990; Breitenbach-Dorfer et al., 1992). Clearly, more research is needed to discriminate between these hypotheses.
References
Barton, N. H. and Slatkin, M. (1986). A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity, 56: 409–415.
Bergmann, F. (1991). Causes and consequences of species-specific genetic variation patterns in European forest tree species: examples with Norway spruce and silver fir. In: Müller-Starck, G. and Ziehe, M. (eds) Genetic Variation in European Populations of Forest Trees, pp. 192–204. Sauerländer's Verlag, Frankfurt.
Breitenbach-Dorfer, M., Pinsker, W., Hacker, R. and Müller, F. (1992). Clone identification and clinal allozyme variation in populations of Abies alba from the Eastern Alps. Pl Syst Evol, 181: 109–120.
Cavalli-Sforza, L. L. and Edwards, A. W. F. (1967). Phylogenetic analysis: models and estimation procedures. Evolution, 21: 550–570.
Clapham, A. R., Tutin, T. G. and Warburg, E. F. (1962). Flora of the British Isles, 2nd edn. Cambridge University Press, Cambridge.
Comps, B., Thiébaut, B., Paule, L., Merzeau, D. and Letouzey, J. (1990). Allozymic variability in beechwoods (Fagus sylvatica L.) over central Europe: spatial differentiation among and within populations. Heredity, 65: 407–417.
Comps, B., Thiébaut, B. and Merzeau, D. (1991). Genetic variation in European beech stands. In: Müller-Starck, G. and Ziehe, M. (eds) Genetic Variation in European Populations of Forest Trees, pp. 110–124. Sauerländer's Verlag, Frankfurt.
Giles, B. E. and Goudet, J. (1997). Genetic differentiation in Silene dioica metapopulations: estimation of spatiotemporal effects in a successional plant species. Am Nat, 149: 507–526.
Grime, J. P., Hodgson, J. G. and Hunt, R. (1988). Comparative Plant Ecology. A Functional Approach to Common British Species. Unwin Hyman, London.
Hamrick, J. L. and Godt, M. J. W. (1989). Allozyme diversity in plant species. In: Brown, A. H. D., Clegg, M. T., Kahler, A. L. and Weir, B. S. (eds) Plant Population Genetics, Breeding and Genetic Resources, pp. 43–63. Sinauer Associates, Sunderland, MA.
Hamrick, J. L., Godt, M. J. W. and Sherman-Broyles, S. L. (1992). Factors influencing genetic diversity in woody plant species. New Forests, 6: 95–124.
Hamrick, J. L., Godt, M. J. W. and Sherman-Broyles, S. L. (1995). Gene flow among plant populations: evidence from genetic markers. In: Hoch, P. C. and Stephenson, A. G. (eds) Experimental and Molecular Approaches to Plant Biosystematics, pp. 215–232. Missouri Botanical Garden, MO.
Kirby, G. C. (1975). Heterozygote frequencies in small subpopulations. Theor Pop Biol, 8: 31–48.
Kullman, L. (1986). Temporal and spatial aspects of subalpine populations of Sorbus aucuparia in Sweden. Ann Bot Fenn, 23: 267–275.
Lagercrantz, U. and Ryman, N. (1990). Genetic structure of Norway spruce (Picea abies): concordance of morphological and allozymic variation. Evolution, 44: 38–53.
Levin, D. A. and Kerster, H. W. (1974). Gene flow in seed plants. Evol Biol, 7: 139–220.
McCauley, D. E., Raveill, J. and Antonovics, J. (1995). Local founding events as determinants of genetic structure in a plant metapopulation. Heredity, 75: 630–636.
Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA, 70: 3321–3323.
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89: 583–590.
Nei, M. and Chesser, R. K. (1983). Estimation of fixation indices and diversities. Ann Hum Genet, 47: 253–259.
Rameau, J. C., Mansion, D. and Dumé, G. (1989). Flore Forestière Française. Institut pour le développement forestier, Paris.
Raspé, O. (1998). Biologie de la reproduction et variation génétique d'un arbre entomophile: Sorbus aucuparia L. (Rosaceae: Maloideae). Ph.D. Thesis, Catholic University of Louvain, Louvain-la-Neuve, Belgique.
Raspé, O., Jacquemart, A.-L. and Desloover, J. R. (1998). Isozymes in Sorbus aucuparia (Rosaceae: Maloideae): genetic analysis and evolutionary significance of zymograms. Int J Plant Sci, 159: 627–636.
Raymond, M. and Rousset, F. (1995a). GENEPOP(version 1.2): population genetics software for exact tests and ecumenicism. J Hered, 86: 248–249.
Raymond, M. and Rousset, F. (1995b). An exact test for population differentiation. Evolution, 49: 1280–1283.
Reynolds, J., Weir, B. S. and Cockerham, C. C. (1983). Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics, 105: 767–779.
Schoen, D. J. and Brown, A. H. D. (1991). Intraspecific variation in population gene diversity and effective population size correlates with the mating system in plants. Proc Natl Acad Sci USA, 88: 4494–4497.
Slatkin, M. (1985). Rare alleles as indicators of gene flow. Evolution, 39: 52–65.
Slatkin, M. (1987). Gene flow and the geographic structure of natural populations. Science, 236: 787–792.
Slatkin, M. (1993). Isolation by distance in equilibrium and non-equilibrium populations. Evolution, 47: 264–279.
Snow, B. and Snow, D. (1988). Birds and Berries. Poyser, London.
Sokal, R. R. and Rohlf, F. J. (1995). Biometry, 3rd edn. W.H. Freeman & Co., New York, NY.
Swofford, D. L. and Selander, R. B. (1981). BIOSYS-1: a Fortran program for the comprehensive analysis of data in population genetics and systematics. J Hered, 72: 281–283.
Vekemans, X. and Lefèbvre, C. (1997). On the evolution of heavy metal tolerant populations in Armeria maritima: evidence from allozyme variation and reproductive barriers. J Evol Biol, 10: 175–191.
Wade, M. J. and McCauley, D. E. (1988). Extinction and recolonization: their effects on the genetic differentiation of local populations. Evolution, 42: 995–1005.
Weir, B. S. and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution, 38: 1358–1370.
Whitlock, M. C. and McCauley, D. E. (1990). Some population genetic consequences of colony formation and extinction: genetic correlations within founding groups. Evolution, 44: 1717–1724.
Zanetto, A. and Kremer, A. (1995). Geographical structure of gene diversity in Quercus petraea (Matt.) Liebl. I. Monolocus patterns of variation. Heredity, 75: 506–517.
Acknowledgements
We would like to thank P. Vakkari, P. Lahdesmaki, A. Weiserbs and P. Fayt who provided Finnish samples, P. Coquillard, J. Gueugnot and G. Mahy for their help in sampling French populations. We are also grateful to J. Thompson and X. Vekemans for their comments and suggestions on the first draft of the manuscript. The assistance of C. No¨el and L. Dhondt was greatly appreciated. This study was financially supported by the catholic University of Louvain (FDS grant no. 629108) and the Belgian National Fund for Scientific Research (FNRS) where O. Raspé is a research assistant and A.-L. Jacquemart is a research associate.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Raspé, O., Jacquemart, AL. Allozyme diversity and genetic structure of European populations of Sorbus aucuparia L. (Rosaceae: Maloideae). Heredity 81, 537–545 (1998). https://doi.org/10.1046/j.1365-2540.1998.00416.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.1998.00416.x
