
Abstract
Genetic diversity in the nuclear genome of Acacia mangium was estimated using 57 anonymous RFLP loci for 10 individuals from each of 10 natural populations representing the geographical range of the species. The level of genetic diversity varied significantly among the populations, ranging from HE=0.01 on the island of Ceram to HE=0.21 in Muting, New Guinea. The small, geographically isolated populations of Daintree, Townsville, Ceram and Sidei had low levels of diversity (HE=0.01–0.09) whereas the large New Guinea and the Cape York Peninsula populations had higher levels of diversity (HE=0.16–0.21). There was evidence of genetic differentiation between populations (θ=0.35), 75% of which was attributable to differences between four geographical regions of Cape York and Daintree–Townsville in Australia, New Guinea and Ceram–Sidei. This may reflect restrictions to gene flow associated with past expansions and contractions of rainforests which occurred with climate and sea level changes. The level of variation detected with RFLPs was higher than previously detected with allozymes, supporting speculation that allozyme coding regions are more likely to be constrained by selection than noncoding regions. The distribution of variation within and among populations was consistent for the two marker types.
Similar content being viewed by others
Introduction
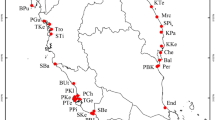
Acacia mangium Willd. is one of only nine acacias with distributions extending from Australia to New Guinea (Skelton, 1986). It typically occupies a narrow ecotone between rainforest and sclerophyllous forest in the seasonally dry tropics. Within Australia, A. mangium has a discontinuous distribution, generally occurring at altitudes less than 100 m, on rainforest margins from the central Queensland coast (Townsville) to the Cape York Peninsula. In the southern lowlands of New Guinea, populations are more extensive and are associated with monsoon vine forests and open savannah woodland. Isolated populations occur in northern Irian Jaya (Sidei) and the Moluccas (Sula, Ceram and Aru) (Fig. 1). Acacia mangium has become an important forest tree in south-east Asia where over 500000 ha of plantations have been established for pulp and paper production.

The current known natural distribution of Acacia mangium and location of seed collection sites.
Studies of genetic diversity in A. mangium revealed levels of allozyme diversity (HT=0.025, Moran et al., 1989a; HT=0.070, Khasa et al., 1994) that were too low to allow investigation of the breeding system or to estimate gene flow among populations. Standard electrophoretic techniques do not detect all amino acid substitutions and may therefore underestimate genetic variability when compared with DNA markers (Lewontin, 1985). Comparative studies of allozymes and DNA markers from the nuclear genome of forest trees have revealed similar or higher estimates of genetic diversity. For example, Pinus resinosa (Mosseler et al., 1992) and Thuja plicata (Glaubitz, 1995) have low levels of allozyme variation and low RAPD and RFLP variation, respectively, whereas in Eucalyptus nitens (Byrne et al., 1998) and Pinus sylvestris (Karhu et al., 1996) RFLP diversity was higher than allozyme diversity. Differences have also been reported in the distribution of variation using different markers. Allozyme frequency profiles can result from active selection rather than random genetic drift (Lewontin, 1985; Bush & Smouse, 1992) and may reveal patterns of variation different from DNA markers because of different selective forces operating on the different marker loci (Mitton, 1994). In Beta vulgaris a higher proportion of variation was attributed to differences among populations based on RFLP data than on allozymes, indicating that uniform balancing selection may operate to maintain approximately equal allele frequencies among populations at allozyme loci (Raybould et al., 1997).
To investigate whether DNA markers could provide additional information on the patterns of genetic diversity and gene flow within and among natural populations of A. mangium, populations from throughout the geographical range of the species were surveyed using RFLP markers.
Materials and methods
Plant material
Seed collected from 10 individuals in each of eight natural populations and bulked seedlots from a further two populations were used to represent the species' geographical range (Fig. 1). The location and details of seedlots are listed in Table 1. All trees sampled were mature individuals, at a minimum distance of 100 m apart. One seed from each individual and 10 seeds from bulked seedlots were germinated, grown in the glasshouse, and leaves collected for DNA extraction.
DNA isolation
Total genomic DNA was extracted from 5 to 10 g of leaves using a modified CTAB procedure (Murray & Thompson, 1980). The concentration of CTAB in the extraction buffer was increased to 3% and NaCl to 1.8 M to minimize the precipitation of polysaccharides with the DNA. For samples which still contained high levels of polysacchides, DNA was digested with ‘Caylase’ following procedures described by Rether et al. (1993).
Library construction
A random genomic library was constructed from A. mangium total DNA (CSIRO seedlot 13229 — Claudie River) using standard methods (Sambrook et al., 1989). PstI-restricted DNA fragments ranging from 0.5 to 2.3 kb were ligated into dephosphorylated pUC19 and transformed into E. coli. Transformant colonies were selected for the presence of recombinant plasmids using IPTG and Xgal and screened to remove plasmids containing highly repetitive DNA. Dilutions of overnight cultures of bacterial colonies (1:50 in d H20) were denatured and stored at −20°C for PCR amplification.
RFLP procedures
DNA samples from the 100 A. mangium individuals were digested with DraI, EcoRI, BglII and HindIII, electrophoresed and transferred to nylon membranes by capillary blotting. Probes were prepared by PCR amplification of inserts and labelled with 32P-dCTP using the random priming method (Feinberg & Vogelstein, 1983). Hybridization and autoradiography followed Byrne et al. (1993) except that posthybridization washes were in 2×SSC, 0.1% SDS.
Probe selection
DNA from one individual in each of the 10 natural populations was digested with each of four restriction enzymes (DraI, EcoRI, BglII and HindIII). One enzyme was selected for each of 50 probes which showed banding patterns that were consistent with simple Mendelian inheritance.
Data analysis
Bands detected by a probe were interpreted as alleles at a genetic locus. Alleles were numbered consecutively according to size and the largest sized fragment designated allele 1. Allele frequencies were calculated using BIOSYS-1 (Swofford & Selander, 1989) and used to estimate the following measures of genetic diversity: mean number of alleles per locus (A); mean percentage of polymorphic loci (P); observed heterozygosity (HO); and Hardy–Weinberg expected panmictic heterozygosity (HE). The inbreeding coefficients (f), total inbreeding coefficient (F) and coancestry coefficient (θ) were calculated using the GDA program (Lewis & Zaykin, 1997), based on the formulae in Weir (1996). Deviations of F, f and θ from zero were tested using bootstrapping (1000 samples) to estimate confidence intervals. To allow comparison with published estimates from allozyme data the relative degree of differentiation between populations (GST) (Nei, 1973) was also calculated.
A hierarchical cluster analysis (UPGMA) was performed using Nei's (1978) unbiased genetic distances and standard errors calculated using the method of Ritland (1989). The distribution of genetic variation among four geographical regions was examined by calculating genetic diversity of each region (HR) and the mean genetic differentiation within and between regions (DSR and DRT, respectively). The proportion of variation between populations which was attributable to differences between regions (DRT/HT) could then be determined using the formula

Two indirect measures of gene flow were calculated. The mean number of migrants exchanged between populations per generation (Nm) was estimated from the mean frequency of alleles found in only one population [private alleles p (1) ]. The formula used for 10 individuals per population was (Slatkin & Barton, 1989)

Results were compared with estimates using Wright's FST (equated with θ in our calculations) and the formula Nm=(1/FST−1)/4 (Slatkin, 1987).
Estimates of Nm are based on the assumption of random mating and may be biased given evidence of inbreeding. The data set was therefore subdivided and gene flow examined between regions within which there was no evidence of nonrandom mating (i.e. f not significant).
Results
Level of genetic variation
The bands revealed by the 50 probes were genetically interpreted as alleles at 57 putative Mendelian loci. Forty-six loci were polymorphic in one or more of the 10 natural populations. Allele frequencies were calculated from all loci (data available from the authors) and used to estimate genetic diversity for the 10 populations (Table 2). The highest measures of genetic variation (allelic diversity, number of polymorphic loci and heterozygosity) were observed in the New Guinea populations (Pongaki, Makapa, Muting and Lake Murray). Within Australia, diversity declined from north to south with the Townsville and Daintree populations having approximately half the genetic variation detected in the Cape York populations (Claudie River and Captain Billy Road). The lowest level of genetic variation was recorded in Sidei and Ceram.
There was evidence of significant levels of inbreeding (Table 2), with an excess of homozygotes in the Townsville, Daintree and Claudie River populations. The distribution of alleles indicated substructuring in the Townsville population with four individuals being homozygous for a different allele than the other six individuals at three loci. No heterozygotes were detected at these loci.
Distribution of genetic variation
The level of differentiation among populations was high (Table 3) and estimates of confidence intervals for θ indicate that significant divergence has occurred between the populations. This reflects differences in the identities of the most common allele between populations and the localized occurrence of rare alleles. At 18 of the 46 variable loci, different alleles were most common in different populations. Rare alleles were concentrated in the New Guinea and Cape York populations and were not detected in the Townsville, Sidei and Ceram populations.
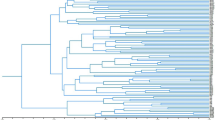
Nei's genetic distances between populations ranged from 0.009 to 0.189 with a mean of 0.083. This was an order of magnitude higher than previous estimates from allozymes (Table 4) and was higher than the mean of 0.045 reported from allozyme data for conspecific populations (Gottlieb, 1977; Crawford, 1989). Cluster analysis grouped populations into four distinct regions which correspond with geographical discontinuities in the species distribution (Fig. 2). The greatest genetic distances separated the geographically isolated Ceram and Sidei populations from all other populations. Differences between geographical regions accounted for 75% of the variation between populations. There was evidence of significantly higher levels of divergence between populations in Australia (GSR=22%) than New Guinea (5%) (Table 5).

Cluster analysis of populations of Acacia mangium based on Nei's (1978) unbiased genetic distance and the UPGMA algorithm. (Cophenetic correlation=0.909.) A cluster is statistically significant if the standard error bar, depicted as a broad line, is less than half the branch length.
Gene flow
The mean number of migrants exchanged among populations per generation, estimated using the private alleles method, (Nm=0.86) indicates that gene flow has been below the level required to prevent genetic differentiation through the effect of genetic drift alone. Private alleles were concentrated in the New Guinea populations. Of a total of 31 private alleles, four were detected in Pongaki, seven in Makapa, eight in Muting, five in Lake Murray, six in Captain Billy Road and one in Sidei. None was detected in Townsville, Daintree, Claudie River or Ceram. Estimates of gene flow using Wright's FST method, which reflects differences in both common and rare alleles, were lower (Nm=0.46).
Comparison of gene flow between geographical regions (Table 6) showed evidence of restrictions to gene flow between the Cape York and southern Queensland populations and between Sidei and Ceram and all other populations. However, it appears that despite the geographical barrier of Torres Strait there has been sufficient gene flow between New Guinea and Australia to prevent differentiation among these regions.
Discussion
The level of genetic diversity within populations of A. mangium varied throughout the geographical range of the species and was consistent with differences in their size and degree of geographical isolation. Populations in New Guinea which are large and widespread were characterized by relatively high levels of genetic diversity, whereas those in the Daintree–Townsville region, which tend to be small and widely scattered (Harwood & Williams, 1992), had low levels of diversity. The decline in genetic diversity from north to south and the higher differentiation between populations in Australia compared with New Guinea may reflect decreasing opportunities for gene flow between more isolated populations of smaller size. Evidence of substructuring in the Townsville population also indicates limited gene flow which increases the opportunity for consanguineous matings.
The extremely low levels of genetic diversity in the geographically isolated populations of Sidei and Ceram suggest that these populations have been founded on a narrow genetic base or have experienced a decline in diversity because of isolation and small size. Bottlenecks associated with long-distance founding events, the absence of repeated migration as a source of genetic enrichment and the possibility of novel selection pressures in new environments are all likely to lead to a loss of genetic variation during colonizing events, particularly those that involve long-distance migration (Barrett & Husband, 1989). The large genetic distance between Sidei, Ceram and other natural populations argues against relatively recent establishment from those sampled in Australia or New Guinea. Sidei also contained a common allele that was not detected in any other population sampled, raising questions over possible source populations. The populations on Aru and Sula have not been genetically assessed.
Differentiation among populations
The level of differentiation among populations (GST=0.331) was high compared to the mean GST of 0.102 estimated from allozyme data for 73 woody angiosperms (Hamrick, 1992); 0.264 for Australian forest trees (Moran, 1992) and 0.162 estimated for Eucalyptus nitens using RFLPs (Byrne et al., 1998). It was within the range of 0.00–0.384 reported for 13 species of Juglans (Fjellstrom & Parfitt, 1994) and was similar to that previously recorded for A. mangium from allozyme data (GST=0.311) by Moran et al. (1989a). A lower GST estimate was reported for A. mangium by Khasa et al. (1994) (Table 4), but this may reflect the inclusion of introduced populations which were of unknown and possibly mixed origins. Although the genetic distances between natural population of A. mangium estimated from the RFLP data were an order of magnitude higher than previously recorded from allozymes, the distribution of variation within and between populations was similar for both marker types.
The relatively high level of divergence among populations of A. mangium may reflect both barriers to gene flow and diversifying selection. The current distribution of A. mangium occurs entirely within the Australian plate. The islands of Ceram, Aru and Timor occupy extreme ends of a single tectonic unit, rifted from north-western Australia in the Jurassic (Michaux, 1991). The distribution of the species, together with those of six other acacias from the Juliflorae which occur in New Guinea and Australia, suggest that the group was more widespread prior to sea level rises which resulted in the Torres Strait reaching its current extent 6500 years ago. The occurrence of A. mangium at low elevations in coastal or riverine environments would have increased its susceptibility to sea level changes. Sea level falls which accompanied glaciation during the Quaternary period would have favoured its dispersal whereas rises would have reduced its geographical range.
Radiocarbon dating of charcoal fragments under rainforest has shown that much of the area currently covered by rainforest in north-eastern Australia was dominated by fire-prone eucalypt woodland between 27000 and 3500 years ago (Hopkins et al., 1993). In the late Pleistocene the distribution of rainforest appears to have been a highly fragmented version of that which exists today, persisting in fire-proof topographic and edaphic locations. Existing rainforests are therefore relatively recent, having reinvaded from refugia. Palynological evidence also indicates that decreases in rainfall and temperature which occurred at the height of the glaciation ≈18000 BP (Kershaw, 1989) favoured the expansion of Eucalyptus and rainforest displacement. During this time A. mangium would have been restricted to moister refugia and the edges of mesic forests in New Guinea. The relatively low diversity and evidence of substructuring in the Townsville and Daintree populations are consistent with founder effects associated with the contraction into and expansion from refugia. The lower level of gene flow among the Australian populations than between Daintree–Townsville and New Guinea could be explained if diversifying selection occurred during and following the species expansion from refugia.
Inbreeding
Species with wide geographical ranges generally maintain moderately high levels of gene flow resulting in low levels of divergence among populations if they have outcrossing mating systems (Hamrick, 1992). There have been no published accounts of the breeding system in A. mangium, but Australian acacias appear to be highly outcrossing (Moran et al., 1989b; Muona et al., 1991; Khasa et al., 1993) with self-incompatibility mechanisms (Kenrick et al., 1986). Although the mean population fixation index for A. mangium calculated from the RFLP data was close to zero, there was evidence of significant inbreeding in the Australian populations (f=0.17). Either the effects of climate and sea level change were more severe on these populations (Boland et al., 1990) or inbreeding may be associated with fragmentation resulting from relatively recent changes in land use. The Daintree and Townsville populations have been affected by rural and urban development resulting in barriers to gene flow which would facilitate an increase in consanguineous matings. In contrast, there was no evidence of inbreeding in the Captain Billy Road and New Guinea populations which have been less affected by changes in land use.
Comparison of markers
The level of genetic variation detected in A. mangium using RFLP markers was three to eight times higher than previously detected using allozymes (Table 4). In contrast, several other species with low allozyme variation also have low RAPD variation, for example Amentotaxus formosana (Wang et al., 1996) and Pinus resinosa (red pine) (Mosseler et al., 1992) and low RFLP variation, for example Thuja plicata (western red cedar) (Glaubitz, 1995) and Populus grandidentata (bigtooth aspen) (Liu & Furnier, 1993). The low RFLP variation may, however, be associated with the use of a single restriction enzyme.
In A. mangium differences in sampling strategies may have contributed to the lower level of diversity in the allozyme surveys which focused on populations in Australia, with only two New Guinea populations represented. However, even the more variable populations in New Guinea had considerably lower allozyme variation (HE=0.028–0.042) than RFLP variation (HE=0.183–0.211). The detection of higher levels of variation using RFLPs may reflect fundamental differences between the marker types. Allozymes are gene products and reveal nonsilent differences in protein-coding genes whereas RFLPs reveal differences directly at the DNA level in both coding and noncoding regions (Clegg, 1989). The probes used in this study, being derived from a genomic rather than a cDNA library, are more likely to uncover variation in noncoding regions which accumulate mutations more rapidly than coding regions (Devey et al., 1991).
The higher level of variation detected with RFLPs in A. mangium may indicate selective constraints on protein-coding genes in this species. This would lead to a lower rate of (nonsilent) mutation at the loci relative to the rate of (silent and nonsilent) mutation at nuclear RFLP loci. It is conceivable that allozyme and RFLP variation have been reduced by a past population bottleneck but that RFLPs have since recovered some of their variation because of their higher mutation rate. Species that have low variation at both allozymes and DNA markers may have experienced more recent and severe bottlenecks than A. mangium.
Alternatively, the recovery of DNA variation in A. mangium and not in red pine, poplar and western red cedar may be associated with differences in their breeding systems. High rates of selfing have been reported in western red cedar (s=0.68) (El-Kassaby et al., 1994) and poplars can reproduce clonally (Cheliak & Dancik, 1982). Although there have been no direct estimates of outcrossing in red pine, high self-pollination has been observed in isolated red pine trees (Mosseler et al., 1992). Lower RFLP diversity was recorded in the populations of A. mangium with evidence of higher levels of inbreeding. This suggests that species or populations with high levels of inbreeding or selfing maintain low levels of both RFLP and allozyme variation following a bottleneck whereas outcrossing facilitates the recovery of RFLP variation.
The pattern of RFLP variation in A. mangium is consistent with the species previously having a more widespread distribution. Fragmentation of populations has been greatest in the southern portion of its range resulting in a decline in genetic diversity and increase in inbreeding. The extremely low genetic diversity in Sidei and Ceram is consistent with a bottleneck associated with long-distance founding events or population contraction because of long-term isolation. Although RFLPs detected a higher level of genetic diversity than allozymes, the distribution of variation within and between populations was similar. The greatest discrepancy between markers was in estimates of genetic distance which were an order of magnitude higher for RFLPs. This could reflect uniform balancing selective pressure on protein-coding genes in a species adapted to a relatively narrow ecological niche and a higher rate of mutation at RFLP loci.
References
Barrett, S. C. H. and Husband, B. C. (1989). The genetics of plant migration and colonisation. In: Brown, A. H. D., Clegg, M. T., Kahler, A. L. and Weir, B. S. (eds) Plant Population Genetics, Breeding and Genetic Resources, pp. 254–277. Sinauer Associates, Sunderland, MA.
Boland, D. J., Pinyopusarerk, K., McDonald, M. W., Jovanovic, T. and Booth, T. H. (1990). The habitat of Acacia auriculiformis and probable factors associated with its distribution. J Trop For Sci, 3: 159–180.
Bush, R. M. and Smouse, P. E. (1992). Evidence for the adaptive significance of allozymes in forest trees. New Forests, 6: 179–196.
Byrne, M., Moran, G. F. and Tibbits, W. (1993). Restriction map and maternal inheritance of chloroplast DNA in Eucalyptus nitens. J Hered, 84: 218–220.
Byrne, M., Parish, T. L. and Moran, G. F. (1998). Nuclear RFLP diversity in Eucalyptus nitens. Heredity, 81: 214–224.
Cheliak, W. M. and Dancik, B. P. (1982). Genetic diversity of natural populations of a clone-forming tree Populus tremuloides. Can J Genet Cytol, 24: 611–616.
Clegg, M. T. (1989). Molecular diversity in plant populations. In: Brown, A. H. D., Clegg, M. T., Kahler, A. L. and Weir, B. S. (eds) Plant Population Genetics, Breeding and Genetic Resources, pp. 98–115. Sinauer Associates, Sunderland, MA.
Crawford, D. J. (1989). Enzyme electrophoresis and plant systematics. In: Soltis, D. E. and Soltis, P. S. (eds) Isozymes in Plant Biology, pp. 146–164. Chapman and Hall, London.
Devey, M. E., Jermstad, K. D., Tauer, C. G. and Neale, D. B. (1991). Inheritance of RFLP loci in a loblolly pine three-generation pedigree. Theor Appl Genet, 83: 238–242.
El-Kassaby, Y. A., Russell, J. and Ritland, K. (1994). Mixed mating in an experimental population of western red cedar Thuja plicata. J Hered, 85: 227–231.
Feinberg, A. P. and Vogelstein, B. (1983). A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Analyt Biochem, 132: 6–13.
Fjellstrom, R. G. and Parfitt, D. E. (1994). Walnut (Juglans spp.) genetic diversity determined by restriction fragment length polymorphisms. Genome, 37: 690–700.
Glaubitz, J. C. (1995). Applications of Molecular Markers to Forest Genetics: Genetic Diversity, Genetic Linkage Mapping and Gene Expression. Ph.D. Thesis, Department of Forest Sciences and the Biotechnology Laboratory, University of British Columbia, Vancouver, BC.
Gottlieb, L. D. (1977). Electrophoretic evidence and plant systematics. Ann Mo Bot Gard, 64: 161–180.
Hamrick, J. L. (1992). Distribution of genetic diversity in tropical tree populations: Implications for the conservation of genetic resources. In: Resolving Tropical Forest Resource Concerns Through Tree Improvement, Gene Conservation and Domestication of New Species, pp. 74–82. IUFRO Conference Proceedings, Colombia.
Harwood, C. E. and Williams, E. R. (1992). A review of provenance variation in growth of Acacia mangium In: Carron, L. T. and Aken, K. M. (eds) Breeding Technologies for Tropical Acacias, pp. 22–30. ACIAR Proceedings no. 37, ACIAR, Canberra.
Hopkins, M. S., Ash, J., Graham, A. W., Head, J. and Hewett, R. K. (1993). Charcoal evidence of the spatial extent of the Eucalyptus woodland expansions and rainforest contractions in North Queensland during the late Pleistocene. J Biogeogr, 20: 357–372.
Karhu, A., Hurme, P., Karjalainen, M., Karvonen, P., Karkkainen, K., Neale, D. et al. (1996). Do molecular markers reflect patterns of differentiation in adaptive traits in conifers? Theor Appl Genet, 93: 215–221.
Kenrick, J., Kaul, V. and Williams, E. G. (1986). Self-incompatibility in Acacia retinodes: site of pollen tube arrest is the nucellus. Planta, 169: 245–250.
Kershaw, A. P. (1989). Was there a ‘Great Australian Arid Period’? Search, 20: 89–92.
Khasa, P. D., Cheliak, W. M. and Bousquet, J. (1993). Mating system of Racosperma auriculiforme in a seed production area in Zaire. Can J Bot, 71: 779–785.
Khasa, P. D., Cheliak, W. M. and Bousquet, J. (1994). Genetic variation in 26 populations of Racosperma auriculiforme and Racosperma mangium using allozymes. Can J Forest Res, 24: 1123–1132.
Lewis, P. O. and Zaykin, D. (1997). Genetic Data Analysis: Computer program for the analysis of allelic data Version 1.0. Free program distributed by the authors over the internet from the GDA Home Page at http://chee.unm.edu/gda/.
Lewontin, R. C. (1985). Population genetics. Ann Rev Genet, 19: 81–102.
Liu, Z. and Furnier, G. R. (1993). Comparison of allozyme, RFLP, and RAPD markers for revealing genetic variation within and between trembling aspen and bigtooth aspen. Theor Appl Genet, 87: 97–105.
Michaux, B. (1991). Distributional patterns and tectonic development in Indonesia: Wallace reinterpreted. Aust Syst Bot, 4: 25–36.
Mitton, J. B. (1994). Molecular approaches to population biology. Ann Rev Ecol Syst, 25: 45–69.
Moran, G. F. (1992). Patterns of genetic diversity in Australian tree species. New Forests, 6: 49–66.
Moran, G. F., Muona, O. and Bell, J. C. (1989a). Acacia mangium: a tropical forest tree of the coastal lowlands with low genetic diversity. Evolution, 43: 231–235.
Moran, G. F., Muona, O. and Bell, J. C. (1989b). Breeding systems and genetic diversity in Acacia auriculiformis and A. crassicarpa. Biotropica, 21: 250–256.
Mosseler, A., Egger, K. N. and Hughes, G. A. (1992). Low levels of genetic diversity in red pine confirmed by random amplified polymorphic DNA markers. Can J Forest Res, 22: 1332–1337.
Muona, O., Moran, G. F. and Bell, J. C. (1991). Hierarchical patterns of correlated mating in Acacia melanoxylon. Genetics, 127: 619–626.
Murray, M. G. and Thompson, W. F. (1980). Rapid isolation of high molecular weight plant DNA. Nucl Acids Res, 8: 4321–4325.
Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci, USA, 70: 3321–3323.
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89: 583–590.
Raybould, A. F., Mogg, R. J. and Gliddon, C. J. (1997). The genetic structure of Beta vulgaris ssp. maritima (sea beet) populations. II. Differences in gene flow estimated from RFLP and isozyme loci are habitat-specific. Heredity, 78: 532–538.
Rether, B., Delmas, G. and Laouedj, A. (1993). Isolation of polysaccharide-free DNA from plants. Plant Mol Biol Rep, 11: 333–337.
Ritland, K. (1989). Genetic differentiation, diversity and inbreeding in the mountain monkey flower (Mimulus caespitosus) of the Washington Cascades. Can J Bot, 67: 2017–2024.
Sambrook, J., Fritsch, E. F. and Maniatis, T. (1989). Molecular Cloning. A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, NY.
Skelton, D. J. (1986). Distribution and ecology of Papua New Guinea acacias. In: Turnbull, J. W. (ed.) Australian Acacias in Developing Countries, pp. 38–44. ACIAR Proceedings no. 16, ACIAR, Canberra.
Slatkin, M. (1987). Gene flow and the geographic structure of natural populations. Science, 236: 787–792.
Slatkin, M. and Barton, N. H. (1989). A comparison of three indirect methods for estimating average levels of gene flow. Evolution, 43: 1349–1368.
Swofford, D. L. and Selander, R. B. (1989). BIOSYS-1. A computer program for the analysis of allelic variation in population genetics and biochemical systematics. Release 1.7. University of Illinois, Urbana, IL.
Wang, C. -T., Wang, W. -Y., Chiang, C. -H., Wang, Y. -N. and Lin, T. -P. (1996). Low genetic variation in Amentotaxus formosana Li revealed by isozyme analysis and random amplified polymorphic DNA markers. Heredity, 77: 388–395.
Weir, B. S. (1996). Genetic Data Analysis II. Methods for Discrete Population Genetic Data. Sinauer Associates, Sunderland, MA.
Acknowledgements
We thank the CSIRO Australian Tree Seed Centre (ATSC) for providing seed; J. C. Bell and G. Morosin for technical support; M. McDonald for preparation of Fig. 1; J. Glaubitz and K. Brubaker for comments on the manuscript. Financial support was provided by AusAID.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Butcher, P., Moran, G. & Perkins, H. RFLP diversity in the nuclear genome of Acacia mangium. Heredity 81, 205–213 (1998). https://doi.org/10.1046/j.1365-2540.1998.00392.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.1998.00392.x
Keywords
This article is cited by
-
Detecting mislabeling and identifying unique progeny in Acacia mapping population using SNP markers
Journal of Forestry Research (2017)
-
Genetic variation in natural populations of Acacia visco (Fabaceae) belonging to two sub-regions of Argentina using AFLP
Plant Systematics and Evolution (2016)
-
Genetic variation in growth, stem straightness, pilodyn and dynamic modulus of elasticity in second-generation progeny tests of Acacia mangium at three sites in Vietnam
New Forests (2015)
-
Study of quantitative genetics of gum arabic production complicated by variability in ploidy level of Acacia senegal (L.) Willd
Tree Genetics & Genomes (2015)
-
Genetic entities and hybridisation within the Acacia microbotrya species complex in Western Australia
Tree Genetics & Genomes (2015)
