
Abstract
Aim
To perform a detailed clinical and psychophysical assessment of the members of three British families affected with blue cone monochromatism (BCM), and to determine the molecular basis of disease in these families.
Methods
Affected and unaffected members of three families with BCM were examined clinically and underwent electrophysiological and detailed psychophysical testing. Blood samples were taken for DNA extraction. The strategy for molecular analysis was to amplify the coding regions of the long wavelength-sensitive (L) and middle wavelength-sensitive (M) cone opsin genes and the upstream locus control region by polymerase chain reaction, and to examine these fragments for mutations by direct sequencing.
Results
We have confirmed the reported finding of protan-like D-15 arrangements of patients with BCM. In addition, we have demonstrated that the Mollon–Reffin (MR) Minimal test is a useful colour-discrimination test to aid in the diagnosis of BCM. Affected males were shown to fail the protan and deutan axes, but retained good discrimination on the tritan axis of the MR test, a compelling evidence for residual colour vision in BCM. This residual tritan discrimination was also readily detected with HRR plates. In two families, psychophysical testing demonstrated evidence for progression of disease. In two pedigrees, BCM could be linked to unequal crossovers within the opsin gene array that resulted in a single 5′-L/M-3′ hybrid gene, with an inactivating Cys203Arg mutation. The causative mutations were not identified in the third family.
Conclusions
The MR test is a useful method of detecting BCM across a wide range of age groups; residual tritan colour discrimination is clearly demonstrated and allows BCM to be distinguished from rod monochromatism. BCM is usually classified as a stationary cone dysfunction syndrome; however, two of our families show evidence of progression. This is the first report of progression associated with a genotype consisting of a single 5′-L/M-3′ hybrid gene carrying an inactivating mutation. We have confirmed that the Cys203Arg inactivating mutation is a common sequence change in blue cone monochromats.
Similar content being viewed by others

Introduction
In order to derive colour vision, the normal human visual system compares the rate of quantum catches in three classes of cone, the short (S or blue) wavelength-sensitive, middle (M or green) wavelength-sensitive and long (L or red) wavelength-sensitive cones, which are maximally sensitive to light at 430, 535, and 565 nm, respectively. This triad of cone types provides the physiological substrate for trichromacy.1, 2, 3
Each cone class contains its own visual pigment composed of an opsin protein linked by a Schiff base to the chromophore retinal. In humans, the L- and M-opsins are encoded by genes on the X chromosome, and the S-opsin by a gene located on chromosome 7.4 There exist three types of inherited colour vision deficiency in which vision is dichromatic; each type corresponds to a selective defect in one of the three receptor mechanisms.1, 2 Protanopia and deuteranopia are characterised by defects of the red- and green-sensitive mechanisms, respectively. They are among the common X-linked disorders of colour vision whose association with alterations in the visual pigment gene cluster at Xq28 identified those genes as encoding the red- and green-sensitive visual pigments.4, 5 The wild-type arrangement of the L- and M-opsin genes consists of a head-to-tail tandem array of two or more repeat units of 39 kb on chromosome Xq28 that are 98% identical at the DNA level.5 This high level of identity would appear to pre-dispose the L and M-opsin genes to unequal inter- and intragenic recombination. Transcriptional regulation of the L and M genes is controlled by an upstream locus control region (LCR).6 In contrast, tritanopia is an autosomal dominant disorder that is characterised by a selective defect of the blue-sensitive mechanism and is due to dominant mutations in the S-cone opsin gene.
Blue cone monochromatism (BCM), or X-linked incomplete achromatopsia, is a rare congenital stationary cone dysfunction syndrome, affecting less than 1 in 100 000 individuals, characterised by the absence of L- and M-cone function.2 Thus, blue-cone monochromats possess rod vision and a normal short-wavelength-sensitive cone mechanism. Photopic vision is mediated by the blue cones, and without a comparison between different classes of the cone photoreceptor, affected individuals are reported to have poor colour discrimination. Mutations in the L- and M-opsin gene array that result in the lack of functional L- and M- pigments, and thus inactivate the corresponding cones, have been identified in the majority of BCM cases studied.6, 7
As in rod monochromacy (RM), BCM typically presents with reduced visual acuity, pendular nystagmus, and photophobia. Visual acuity is of the order of 6/24–6/60. Eccentric fixation may be present and myopia is a common finding.8 BCM is distinguished from RM via psychophysical or electrophysiological testing. The photopic electroretinogram (ERG) is profoundly reduced in both, but the S-cone ERG is well preserved in BCM.9 BCM patients have high Farnsworth–Munsell 100-Hue scores, but have fewer errors in the vertical (tritan) axis when compared to RM. At the molecular level, mutation analyses have proved highly efficient at establishing the molecular basis for BCM.6, 7 The mutations in the L- and M-opsin gene array that cause BCM fall into two classes. In the first class, a normal L- and M-opsin gene array is inactivated by a deletion in the LCR, located upstream of the L-opsin gene. A deletion in this region would appear to abolish transcription of all genes in the opsin gene array and therefore inactivates both L- and M-cones.10 In the second class of mutations, the LCR is preserved, but changes within the L- and M-pigment gene array lead to loss of functional pigment production. The most common genotype in this class consists of a single inactivated L/M hybrid gene. The first step in this second mechanism is thought to be unequal crossing over that reduces the number of genes in the array to one, followed by a mutation that inactivates the remaining gene. A frequent inactivating mutation that has been described is the thymine-to-cytosine transition at nucleotide 648, which results in a cysteine-to-arginine substitution at codon 203, a mutation known to disrupt the folding of cone opsin molecules.11
Our aim was to perform a detailed clinical and psychophysical assessment of members of three British families affected with BCM, and subsequently to determine the molecular basis of BCM in these families.
Patients and methods
Affected and unaffected members of three families with BCM were examined clinically and underwent psychophysical and electrophysiological testing. After informed consent was obtained, blood samples were taken, genomic DNA was isolated from whole blood using an extraction kit (Nucleon®Biosciences), and molecular genetic analysis was performed.
Clinical assessment
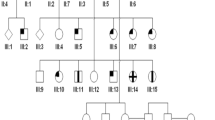
The pedigrees of the families studied can be seen in Figures 1a–c. A full medical and ophthalmic history was taken in all examined members. A full ophthalmological examination was performed. All affected individuals had an ERG performed. Adults had an ERG that conformed to the ISCEV standard, but the children had a modified protocol using skin electrodes. Colour vision testing included the use of Hardy, Rand and Rittler (HRR) plates, SPP2 plates for acquired colour deficiency, Farnsworth–Munsell (FM) 100-hue test, Farnsworth D-15, the Mollon-Reffin (MR) minimal test,12 a computerised colour vision test,13, 14 and anomaloscopy.

Family pedigrees showing X-linked recessive inheritance pattern. Affected males are represented by filled-in symbols, carrier females by a central black dot. (a) Family A: Subject III:2 is deuteranomalous and her father is reported to be red-green colour blind. This is represented by a shaded quadrant. (b) Family B (c) Family C.
The FM 100-hue, Farnsworth D-15 and the MR test were all performed under CIE Standard Illuminant C from a MacBeth Easel lamp. The MR minimal test is a saturation discrimination-type test.12 The caps used in this test are of a similar design to those in the D-15 and FM 100-hue tests. The test features a series of caps that lie along protan, deutan, and tritan lines, respectively. In addition, there is one demonstration cap, which does not lie along a dichromatic confusion axis. The remainder of the caps are all neutral, but have varying lightness (there are a total of nine grey caps). The examiner places one coloured cap among a group of neutral caps and asks the subject to tap the side of the cap that is coloured. The coloured caps vary in their saturation, so the severity of the defect can be assessed. It can be performed by children as young as 5 years.
Molecular genetic analysis
The underlying strategy for molecular analysis was to amplify the coding regions of the L- and M-cone opsin genes and the upstream LCR by PCR, and to examine these fragments for mutations by direct sequencing.
Exonic sequences of the L- and M-genes are 98% identical at the nucleotide level and, although there may be less evolutionary pressure for intronic sequences to remain stable, the introns of the two genes have also remained almost identical (>99.9% identical at the nucleotide level). It was not possible, therefore, to design PCR primers that amplify only L- or only M-opsin exonic fragments from genomic DNA. Consequently, the primer pairs used in this study amplified L and M sequences simultaneously, and individual nucleotide differences between the genes were subsequently utilised for differentiation between the genes in sequence analysis.
(i) PCR amplification of the L- and M- opsin genes: Intronic forward and reverse PCR primers (Table 1) were designed to amplify the LCR, and all six exons of the L- and M-genes from genomic DNA. LCR primers were designed based on the published sequence,10 and encompassed the LCR core sequence. Primer pairs that would co-amplify both L- and M-opsin exonic sequences were designed within each intron approximately 50 bp from the intron–exon junction, in order that the whole of each exon, some flanking DNA, and the splice sites were amplified. The design of these primers was based on the published sequences of the L- and M-opsin genes sequence.5 PCR reactions (50 μl) were performed as follows: 1 × NH4 buffer, 1 mM MgCl2, 200 μM each dNTP, 10 pmols each of sense and antisense primers, 200 ng-1 μg DNA, 1 U BioTaq, X μl dH2O — with the exception that the concentrations of MgCl2 used in LCR, exon 3 and 6 PCR reactions were optimised at 0.5 mM, 0.5 mM and 2 mM respectively — and with annealing at the exon-specific temperatures (Table 1). PCR products were visualised by electrophoresis using low melting temperature agarose gel. Target products were then excised and eluted.
(ii) Mutation analysis of the PCR-amplified exons and LCR was carried out by direct sequencing using a cycle-sequencing kit. After ethanol precipitation, the DNA products were analysed on an ABI 373A automated DNA sequencer and examined for alterations, utilising Sequencing Analysis (ABI PrismTM) and GeneWorksTM software.
Results
Phenotype
Family A ( Figure 1a ) Patient III:3—This 7-year-old boy (proband) was originally seen at 1 year of age. He was found to have horizontal pendular nystagmus, normal fundi, and clear media. A family history of nystagmus, photophobia, and ‘colour blindness’ affecting males of the family, including his grandfather (I:1) and male cousin (III:1), was established. ERG testing revealed absent cone responses, but normal rod responses.
He had a visual acuity of 3/36 in each eye with his myopic correction. Psychophysical testing on the MR test and HRR plates revealed reasonable discrimination only along the tritan axis. On computerised testing, his colour-discrimination ellipses were oriented along the angle that one would expect of someone making colour discriminations based on a comparison of quantum catches in the rods and S-cones.
Patient I:1—This 60-year-old man was found to have a visual acuity of 6/36 in the right eye and 6/60 in the left. He was found to have clear lenses and mild macular retinal pigment epithelial (RPE) changes. As a child, he had obvious nystagmus, but this had improved throughout life, to the point that it was not at all noticeable. He felt that his vision had continued to slowly deteriorate throughout life. Cone ERG responses were absent, but rod responses were normal. On the basis of his results on the HRR plates, the D-15, the MR minimal test, and Nagel anomaloscope, it was concluded that he had no residual colour vision.
His 12-year-old grandson (III:1) had a visual acuity of 6/60 in the right eye and 6/36 in the left and displayed evidence of residual colour discrimination. He had clear media and normal fundi. He showed reasonable discrimination along both the tritan line of MR test and also on the SPP2 tritan plates. He displayed a protan ordering of the D-15. On computerised testing his ellipses were oriented along the angle that is expected for colour discriminations based solely upon a comparison of quantum catches in the rods and the S-cones (Figures 2, 4 and 5).

Patient III:1 (Family A): Colour-discrimination ellipse, oriented along the angle that would be expected with someone making colour discriminations based upon a comparison of quantum catches in the rods and the S-cones.

Normal subject: A typical colour-discrimination ellipse for a normal trichromat. The subject's thresholds are plotted as filled circles in the CIE 1974 chromaticity diagram.

Rod monochromacy: The results of a rod monochromat. Note that the subject fails to discriminate almost all of the most saturated stimuli along each axis tested.
Patient III:2—This 14-year-old daughter of II:2 was asymptomatic. On the Nagel anomaloscope, she was found to be deuteranomalous. On the MR minimal test, she showed good discrimination on the tritan axis, but was badly impaired on both protan and deutan axes.
Patients II:2 and II:3 were both asymptomatic, and on detailed psychophysical testing were found to have normal colour vision. The father of patients III:1 and III:2 was not available for psychophysical testing, but was reported to be ‘colour blind’.
A consistent psychophysical hypothesis from these observations would be that the proband III:3 and III:1 have inherited from their maternal grandfather I:1, via their mothers, an X-chromosome with an altered opsin array that has led to BCM. III:2 has also inherited this altered X-chromosome from her mother, and an X-chromosome that leads to a deuteranomalous phenotype from her father. Since III:3 and III:1 both have some residual colour discrimination and their grandfather has none, it would appear that their condition is not stationary. III:3 and III:1 can be labelled blue-cone monochromats, whereas their grandfather behaves as a rod monochromat, presumably as a result of continued S-cone loss. The lack of colour vision seen in the grandfather is highly unlikely to be due to lenticular changes, since his lenses were found to be clear.
Family B ( Figure 1b ) Patient III:2—This 12-year-old boy (proband) originally presented with pendular nystagmus, poor visual acuity (6/24 in both eyes), and myopia. Ocular media were clear with normal fundi. ERG revealed absent cone but normal rod responses. On psychophysical testing, the anomaloscope and Sloan's test for achromatopsia suggested a rod-dominated spectral sensitivity function. On the D-15, he showed the protan-like pattern reported for BCM by Weis & Biersdorf.15 On the computer test, he failed completely on the protan and deutan lines, but scored nearly normally along the tritan line, which modulates the blue cones. In addition, his ellipses were well aligned to the theoretical S-cone/rod confusion axis, consistent with the mechanism whereby colour discriminations are based upon a comparison between rod and S-cone quantum catches (Figures 3, 4 and 5). Exactly the same pattern was exhibited on the MR minimalist test: he could not find saturated protan and deutan probes among the grey distractors, but could find the least saturated tritan cap. On the SPP2 plates for acquired colour deficiencies, he passed the plates that are failed by those with purely scotopic vision (RM).

Patient III:2 (Family B): Colour-discrimination ellipse, well aligned to the theoretical S-cone/rod confusion axis, and consistent with the mechanism whereby colour discriminations are based on a comparison between rod and S-cone quantum catches.
Patient III:1—This 14-year-old brother of the proband also presented with pendular nystagmus, poor visual acuity (6/24 in the right eye and 6/36 in the left), myopia, and photophobia. Ocular media were clear with normal fundi. ERG revealed absent cone responses but normal rod function. He showed reasonable discrimination only along the tritan axis on HRR testing. He declined further psychophysical testing.
Patient II:1—The 50-year-old mother of III:1 and III:2 was asymptomatic. ERG and colour vision testing was normal.
Patient I:1—The maternal grandfather of the propositus was said to have had poor eyesight since birth and to have always had great problems with colour vision. The grandfather had an elder brother who had also worn glasses and had suffered with poor vision since infancy. Both were deceased at the time of investigation.
Family C ( Figure 1c ) Patient V:4 This 7-year-old boy (proband) originally presented as an infant with nystagmus and photophobia. His current visual acuity was recorded at 6/24 in both eyes. He had clear ocular media with normal fundi. He had a family history of nystagmus, poor visual acuity, and colour vision affecting males of the family including his grandfather (III:10) and uncle (IV:1). ERG revealed absent cone responses but normal rod function. On the D-15, he showed confusions characteristic of congenital red-green deficiency or BCM rather than a scotopic axis (RM). On the MR minimal test, he failed the protan and deutan axes, but discriminated on the tritan axis. On the HRR plates, he passed all the tritan plates and failed all the protan/deutan plates.
Patient V:5 This 6-year-old boy had nystagmus and a visual acuity of 6/36 in the right eye and 6/24 in the left. He had clear ocular media with normal fundi. On the MR test, he failed the protan and deutan axes, but discriminated well on the tritan axis. Computerised testing corroborated these findings; he failed completely on the deutan line and discriminated very poorly on the protan line, which hold almost constant the blue cone signal, but scored almost normally along the tritan line, which modulates the blue cones.
Patient III:10 This 70-year-old grandfather of V:4 and V:5 has had poor vision and nystagmus since childhood. The nystagmus had become less prominent over time. He felt that his vision had gradually deteriorated since childhood. He had early lens opacities with minimal yellowing of the lens and mild macular RPE changes. ERG revealed absent cone responses and normal rod function. On detailed psychophysical testing, there was little evidence of any residual colour vision. On anomaloscopy, he displayed the classical brightness-matching function of an achromat. On the D-15, his responses were anarchic. He failed all plates on the SPP2 series. On the MR minimal test, he failed the protan and deutan axes completely, but did show some residual discrimination on the tritan axis, detecting the most saturated cap.
Patient IV:1 This 50-year-old man who is registered partially sighted was found to have a visual acuity of 6/36 in both eyes, with no nystagmus. He complained of poor vision since childhood and that he had always had trouble with colour vision. He had clear lenses and normal fundi. Little residual colour vision could be detected. On the MR minimal test, he failed completely on all the three axes with both eyes. On anomaloscopy, he behaved like an achromat.
Patients IV:6 and IV:7—Both are asymptomatic and have entirely full colour vision on detailed testing.
Since the grandfather (III:10), uncle (IV:1), and two grandchildren (V:4 and V:5) share the same genotype, the condition appears to be progressive. The children show a residual colour discrimination that is lacking in both older men. There is convincing psychophysical evidence to suggest that the children have functional S-cones. The loss of residual colour vision seen in the two older men in this family is highly unlikely to be due to lenticular changes alone, since the lenses were found to be either clear (IV:1) or only minimally yellow (III:10). A markedly yellow lens would be required to result in any significant loss of colour vision and such yellowing would be unlikely to result in the degree of loss of tritan function seen in this family.
Genotype
A number of PCR primer pairs was used to amplify the X-linked L- and M-opsin genes that underlie the L- and M-pigments and the LCR that controls the opsin gene array. The identification of L- or M-opsin gene depends on a number of sequence differences that are confined to exons 2–5.
Family A Sequence analysis demonstrated the presence of a single M-opsin gene in the array of the affected subjects (Figure 6). However, no further alterations of this gene were identified. Examination of the entire LCR revealed an unaltered sequence. The mutational basis of the colour vision defect in this family remains uncertain.

Comparison of the sequences of exon 5 of the L and M opsin genes with the sequences from II:3 (obligate female carrier) and III:1 (affected male) of family A. Differences between the L and M sequences are indicated by arrowheads. N indicates the presence of both L and M nucleotides at that position in II:3.
Family B Sequence representing only the L gene was present for exons 2–4, while for exon 5 only sequence of the M gene was observed. This is consistent with the presence of only a single 5′-L/M-3′ hybrid gene in affected members of family B in their opsin array. Moreover, alignment of exon 4 of the hybrid gene in these individuals with wild-type exon 4 sequence showed that this opsin gene carries the previously reported T → C nucleotide alteration which encodes the Cys203Arg substitution (Figures 7 and 9). Sequence analysis of obligate carriers demonstrated that both L- and M-opsin genes are present in the opsin gene array and that the T → C transition in exon 4 is also carried in the L-opsin genes in these individuals.

Sequence electropherograms for part of exon 4 from (a) III:1 (affected male) of family B and (b) normal male. Note T → C nucleotide alteration in III:1 which results in the Cys203Arg amino acid substitution.

Diagrammatic representations of the genotypes of Families B and C.
Family C Affected individuals of family C also carry the Cys203Arg mutation within a single 5′-L/M-3′ hybrid gene in the array. In this case, the crossover between the L- and M-genes, which is required for generation of the hybrid gene, occurred within exon 3 (Figures 8 and 9).

Comparisons of the sequences of exon 3 of the L and M opsin genes with the sequences from III:10 (affected) and IV:1 (affected) of family C. Differences between the L and M sequences are indicated by arrowheads.
Discussion
Congenital. achromatopsia can be classified into three categories. (i) Typical rod monochromats (complete achromatopsia) have normal rod function, but lack all sensitivity mediated by cone pigments. (ii) Atypical rod monochromats (incomplete achromatopsia) have some residual cone function and consequently have better visual acuity and some residual colour vision. (iii) Blue-cone monochromats have rod and blue-cone function.
Clinically, individuals in all the three categories share a number of features. They have poor visual acuity, pendular nystagmus, photophobia, and in the majority of cases fundoscopy is unremarkable. Regardless of category, rod monochromats classically have very poor or no colour discrimination, and on a Farnsworth D-15 or FM 100 Hue test show no clear axis, and therefore no preference for confusing any subset of hues. Blue-cone monochromats can be distinguished from both types of rod monochromats by psychophysical threshold testing or electroretinographic studies at different wavelengths.2, 9 Classification can also be aided by family history, because BCM is inherited as an X-linked recessive trait, whereas both types of rod monochromacy are autosomal recessive.
Rod monochromats cannot make colour judgements, but rather will use brightness cues to differentiate between colours. When confronted with tasks where luminance cues cannot be used, these patients perform very poorly indeed (eg FM 100 hue scores are very high, D-15 arrangement patterns are characteristic and ordered along a scotopic axis). This contrasts with blue-cone monochromats who do have access to colour discrimination, though this does depend upon the luminance of the task: at mesopic levels, they have rudimentary dichromatic colour discrimination based upon a comparison of the quantum catches obtained by the rods and the S-cones, with their neutral point lying at about 460–470 nm.16, 17 Hue discrimination is reported to deteriorate with increasing luminance.18 Blue-cone monochromats may be distinguished from rod monochromats by means of colour vision testing: blue-cone monochromats are reported to display fewer errors along the vertical axis in the 100-Hue test (fewer tritan errors), and they may also display protan-like ordering patterns on the D-15.15 In addition, the Berson plates are claimed to provide a good separation of blue cone monochromats from rod monochromats.19, 20 In our case series, we have confirmed the previously reported finding of protan-like patterns on D-15 testing of patients with BCM.15 It should be noted, however, that this finding is not necessarily indicative of colour discrimination, since it is possible that BCM individuals order the chips of this test according to their relative lightness to the S-cone mechanism.21 Relevant to this is our demonstration that affected males also retain reasonable discrimination on the tritan axis of the MR test. Since this test utilises lightness randomisation, it should not be possible for those with BCM to discriminate the target chips by lightness cues. Furthermore, those patients who were examined with a computer-controlled saturation discrimination task displayed discrimination ellipses with major axes well aligned to the theoretical S-cone/rod confusion axis. This provides additional evidence that it is possible to derive colour discrimination at low photopic levels via a comparison of quantum catches in the S-cones and rods, as has been suggested previously.16, 18 It is proposed that the latter two tests, which are suitable for use in children, can be readily employed in probing the differential diagnosis of congenital achromatopsia.
BCM is generally accepted to be a stationary cone dysfunction syndrome, although Fleischman and O'Donnell reported one BCM family with macular atrophy and noted a slight deterioration of visual acuity and colour vision during a 12-year follow-up period, as well as pigmentary changes in the fovea.22 There are two further reports of individuals with BCM displaying a progressive retinal degeneration; in both cases, the genotype consisted of deletion of the LCR.6, 23 In two of our families (A and C), there has also been progression in the severity of the condition. Since, in family A, the children have normal tritan function, and their grandfather has none, it would appear that the condition is not stationary. The two children can be labelled blue-cone monochromats, whereas their grandfather behaves as a rod monochromat, presumably secondary to the death of S-cones. In family C, the grandfather, uncle, and two grandchildren share the same genotype. Since these children show convincing psychophysical evidence that S-cones are still surviving, whereas both older men in this family lack residual colour discrimination, the condition also appears to be progressive in this family. Moreover, the genotype identified in this family of a single 5′-L/M-3′ hybrid gene with an inactivating mutation differs from previous reports of progression, where an LCR deletion was present. There is, therefore, no consistent genotype associated with progression of BCM.
Combined results of previous studies6, 7, 23, 24 provide evidence for the general conclusion, first put forward by Nathans et al,6 that there are different mutational pathways to BCM. The data suggest that 40% of blue-cone monochromat genotypes are the result of a one-step mutational pathway that leads to deletion of the LCR. The remaining 60% of blue-cone monochromat genotypes comprise a heterogeneous group of multi-step pathways. The evidence thus far shows that many of these multi-step pathways produce visual pigment genes that carry the inactivating Cys203Arg mutation. In our study, the genetic mechanism identified was unequal recombination resulting in an array comprising a single 5′-L/M-3′ hybrid gene carrying a Cys203Arg mutation (Figure 9). This genotype has been reported as a cause of BCM in earlier studies.6, 7, 24
The cysteine residue at position 203 of the L/M-opsin protein is highly conserved among all visual pigments and several other seven-transmembrane-spanning receptors.25 This cysteine residue is located in the second extracellular loop of the opsin and, together with a conserved cysteine residue at position 126 in the first extracellular loop, forms a disulphide bond necessary for stabilisation of the tertiary structure of the protein.11 The data presented here confirm the incidence of the Cys203Arg mutation in BCM.
The affected subjects assessed in family A would appear to possess only an M-opsin gene in the visual pigment array, which would be expected to produce dichromacy rather than monochromacy. Sequence analysis failed to reveal a mutation in any of the six opsin gene exons or the LCR. The diagnosis of BCM on clinical and psychophysical grounds is clear in this family, as is the X-linked inheritance; so it would seem most likely that the inactivating mutation in this case has not been identified. Other studies have also failed to detect the genetic alteration that would explain the BCM phenotype in all assessed individuals.7, 26 Indeed, Nathans et al7 reported nine subjects out of 33 individuals with BCM, where the structure of the opsin array did not reveal the genetic mechanism of the phenotype. It is therefore also plausible that further genetic heterogeneity remains to be identified in BCM.
In this report, we present the phenotypic and genotypic findings in a case series of three families with BCM, usually regarded as a stationary disorder. However, two of our families demonstrate progression. Thus, it cannot always be assumed that BCM will remain stationary. In addition, it is not generally recognised that nystagmus associated with BCM may become less prominent over time, as has been observed in our study. We have also demonstrated that the MR minimal test and HRR plates are useful colour-discrimination tests to aid in the diagnosis of BCM. Affected males repeatedly failed the protan and deutan axes, but retained good discrimination on the tritan axis of the MR test, whereas patients with RM perform poorly along all the three axes. It is suggested that this test can be readily employed in probing the differential diagnosis of congenital achromatopsia.
References
Boynton RM . Human Color Vision. Holt, Rinehart & Winston: New York, 1979.
Pokorny J, Smith VC, Verriest G . Congenital color defects. In: Pokorny J, Smith VC, Verriest G, Pinckers AJ (eds) Congenital and Acquired Color Vision Defects. Grune & Stratton: New York, 1979, p 183.
Young T . On the theory of light and colours. Philos Trans R Soc Lond 1802; 92: 12–48.
Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS . Molecular genetics of inherited variation in human color vision. Science 1986; 232: 203–210.
Nathans J, Thomas D, Hogness DS . Molecular genetics of human colour vision: the genes encoding blue, green, and red pigments. Science 1986; 232: 193–202.
Nathans J, Davenport CM, Maumenee IH, Lewis RA, Hejtmancik JF, Litt M et al. Molecular genetics of human blue cone monochromacy. Science 1989; 245: 831–838.
Nathans J, Maumenee IH, Zrenner E, Sadowski B, Sharpe LT, Lewis RA et al. Genetic heterogeneity among blue-cone monochromats. Am J Hum Genet 1993; 53: 987–1000.
Weleber RG, Eisner A . Cone degeneration (‘Bull's eye dystrophies’) and colour vision defects. In: Newsome DA (ed) Retinal Dystrophies and Degenerations. Raven Press: New York, 1988, pp 233–256.
Gouras P, MacKay CJ . Electroretinographic responses of the short-wavelength-sensitive cones. Invest Ophthalmol Vis Sci 1990; 31: 1203–1209.
Wang Y, Macke JP, Merbs SL, Zack DJ, Klaunberg B, Bennett J et al. A locus control region adjacent to the human red and green visual pigment genes. Neuron 1992; 9: 429–440.
Kazmi MA, Sakmar TP, Ostrer H . Mutation of a conserved cysteine in the X-linked cone opsins causes color vision deficiencies by disrupting protein folding and stability. Invest Ophthalmol Vis Sci 1997; 38: 1074–1081.
Mollon JD, Astell S, Reffin JP . A minimalist test of colour vision. In: Drun B, Moreland JD, Serra A (eds) Colour Vision Deficiencies XI. Kluwer Academic Publishers: Dordrecht, 1991, pp 59–67.
Mollon JD, Reffin JP . A computer-controlled colour vision test that combines the principles of Chibret and of Stilling. J Physiol 1989; 414: 5.
Regan BC, Reffin JP, Mollon JD . Luminance noise and the rapid determination of discrimination ellipses in colour deficiency. Vision Res 1994; 34: 1279–1299.
Weis AH, Biersdorf WR . Blue cone monochromatism. J Pediatr Ophthalmol Strabismus 1989; 26: 218–223.
Reitner A, Sharpe LT, Zrenner E . Is colour vision possible with only rods and blue-sensitive cones? Nature 1991; 352: 798–800.
Alpern M, Lee GB, Maaseidvaag F, Miller SS . Colour vision in blue cone monochromacy. J Physiol 1971; 212: 211–233.
Young RS, Price J . Wavelength discrimination deteriorates with illumination in blue cone monochromats. Invest Ophthalmol Vis Sci 1985; 26: 1543–1549.
Berson EL, Sandberg MA, Rosner B, Sullivan PL . Color plates to help identify patients with blue cone monochromatism. Am J Ophthalmol 1983; 95: 741–747.
Haegerstrom-Portnoy G, Schneck ME, Verdon WA, Hewlett SE . Clinical vision characteristics of the congenital achromatopsias. II. Color vision. Optom Vis Sci 1996; 73: 457–465.
Hess RF, Mullen KT, Sharpe LT, Zrenner E . The photoreceptors in atypical achromatopsia. Journal of Physiology 1989; 417: 123–149.
Fleischman JA, O'Donnell Jr FE . Congenital X-linked incomplete achromatopsia. Evidence for slow progression, carrier fundus findings, and possible genetic linkage with glucose-6-phosphate dehydrogenase locus. Arch Ophthalmol 1981; 99: 468–472.
Ayyagari R, Kakuk LE, Coats EL, Bingham EL, Toda Y, Felius J et al. Bilateral macular atrophy in blue cone monochromacy (BCM) with loss of the locus control region (LCR) and part of the red pigment gene. Mol Vis 1999; 5: 13–18.
Reyniers E, Van Thienen MN, Meire F, De Boulle K, Devries K, Kestelijn P et al. Gene conversion between red and defective green opsin gene in blue cone monochromacy. Genomics 1995; 29: 323–328.
Karnik SS, Khorana HG . Assembly of functional rhodopsin requires a disulfide bond between cysteine residues 110 and 187. J Biol Chem 1990; 265: 17520–17524.
Ayyagari R, Kakuk LE, Bingham EL, Szczesny JJ, Kemp J, Toda Y et al. Spectrum of color gene deletions and phenotype in patients with blue cone monochromacy. Hum Genet 2000; 107: 75–82.
Acknowledgements
The work was supported by grants from the British Retinitis Pigmentosa Society and the Guide Dogs for the Blind Association. We are also grateful to the patients who kindly agreed to take part in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Michaelides, M., Johnson, S., Simunovic, M. et al. Blue cone monochromatism: a phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye 19, 2–10 (2005). https://doi.org/10.1038/sj.eye.6701391
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6701391
Keywords
This article is cited by
-
Diagnostic analysis of the highly complex OPN1LW/OPN1MW gene cluster using long-read sequencing and MLPA
npj Genomic Medicine (2022)
-
Novel biallelic loss-of-function KCNV2 variants in cone dystrophy with supernormal rod responses
Documenta Ophthalmologica (2019)
-
Genotype determination of the OPN1LW/OPN1MW genes: novel disease-causing mechanisms in Japanese patients with blue cone monochromacy
Scientific Reports (2018)
-
Gene-based Therapy in a Mouse Model of Blue Cone Monochromacy
Scientific Reports (2017)
-
De novo intrachromosomal gene conversion from OPN1MW to OPN1LW in the male germline results in Blue Cone Monochromacy
Scientific Reports (2016)
