
Abstract
Background:
In a genome-wide scan (GWS) of 175 multiplex prostate cancer (PCa) families from the University of Michigan Prostate Cancer Genetics Project (PCGP), linkage was observed to markers on chromosome 17q21–24, a region that includes two breast cancer susceptibility genes, BRCA1 and BRIP1. BRIP1 is a Fanconi anaemia gene (FANCJ) that interacts with the BRCT domain of BRCA1 and has a role in DNA damage repair. Protein truncating mutations in BRIP1 have been identified in hereditary breast and ovarian cancer families, and a recent report suggested that a recurrent truncating mutation (R798X) may have a role in PCa susceptibility.
Methods:
We examined the role of BRIP1 mutations in hereditary PCa through sequence analysis of 94 individuals from PCGP families showing linkage to 17q.
Results:
A total of 24 single-nucleotide polymorphisms, including 7 missense variants but no protein truncating mutations, were observed.
Conclusion:
The data presented here suggest that BRIP1 truncating mutations are uncommon in PCa cases and do not account for the linkage to chromosome 17q observed in our GWS. Additional investigation is needed to determine the significance, if any, of the observed BRIP1 missense variants in hereditary PCa.
Similar content being viewed by others

Main
Prostate cancer (PCa) is the most commonly diagnosed cancer and the second leading cause of cancer death among American men (Jemal et al, 2009). In addition to age and ancestry, family history is a leading risk factor for developing PCa, suggesting that germline genetic variation has a role in PCa development. However, the number and identities of the genes involved in hereditary PCa are still largely unknown.
There is some evidence to suggest that inherited mutations in breast cancer (BrCa) and ovarian cancer (OvCa) risk genes BRCA1 and BRCA2 also increase the risk of PCa. Several studies have shown that carriers of deleterious, germline mutations in BRCA2 are at an increased risk of developing PCa, especially early-onset PCa (Risch et al, 2001; Thompson and Easton, 2002; Edwards et al, 2003; Kirchhoff et al, 2004; Agalliu et al, 2007). In addition, we recently found that two common BRCA1 single-nucleotide polymorphisms (SNPs) are associated with familial and early-onset PCa (Douglas et al, 2007).
BRIP1, located at 17q22, is a Fanconi anaemia gene (FANCJ) (Litman et al, 2005) that directly interacts with the BRCT domain of BRCA1 (Yu et al, 2003) and has a role in DNA damage repair (Bridge et al, 2005; Litman et al, 2005). BRIP1-deficient cell lines are overly sensitive to DNA crosslinkers cisplatin and mitomycin C (Bridge et al, 2005; Litman et al, 2005), arrest in the S-phase of the cell cycle, and exhibit increased chromosomal instability (Kumaraswamy and Shiekhattar, 2007). Recently, BRIP1 has been identified as a BrCa susceptibility gene. Seal et al (2006) found that carriers of truncating BRIP1 mutations have a relative risk of BrCa of 2.0. The functional relationship between BRIP1 and BRCA1 and their roles in hereditary BrCa make BRIP1 a biologically plausible candidate PCa susceptibility gene.
Prostate cancer linkage studies have identified several areas of the genome that may harbour susceptibility loci, although signals have often been difficult to reproduce between research teams (Schaid, 2004). The University of Michigan Prostate Cancer Genetics Project (PCGP) was the first group to report PCa linkage to chromosome 17q markers (Lange et al, 2003). Chromosome 17q linkage has subsequently turned out to be one of the strongest (Lange et al, 2007) and most reproducible (Gillanders et al, 2004; Xu et al, 2005) linkage signals for hereditary PCa. BRIP1 is ∼20 cM downstream from BRCA1 and both are contained near or within our linkage signal. Previous research from our laboratory failed to identify deleterious BRCA1 truncating mutations in chromosome 17-linked families (Zuhlke et al, 2004). In this study, we examine the role of germline BRIP1 variation in hereditary PCa, focusing on families with linkage evidence to 17q markers.
Methods
Patient selection
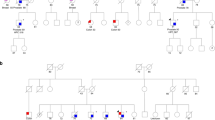
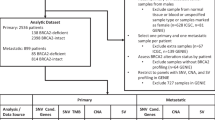
A genome-wide linkage scan was previously performed on 175 families from the PCGP. To be eligible for the scan, families had to meet one of the following criteria: (1) three or more individuals diagnosed with PCa at any age in ⩾3 generations or (2) two or more individuals diagnosed with PCa before the age of 55 years. The most significant evidence for linkage was observed at ∼60 cM on chromosome 17, with 95 families showing some evidence of linkage (non-parametric linkage score >0 at 60.1 cM on chromosome 17, which is the linkage peak from our first genome-wide scan (GWS) (Lange et al, 2003)). One individual was selected from each of 94 linked families and was included in this analysis, including 88 men with PCa and 6 women with BrCa. The selected men were either the youngest PCa case in the family (with DNA available) or an older case in a first- or second-degree relationship with a woman with BrCa. The women selected were all first-degree relatives of at least one man with PCa. The number of BrCa and OvCa cases in each family, as reported in family history surveys, was calculated. PCa cases were confirmed by medical record when possible.
Sequencing
All BRIP1 exons and intron–exon boundaries were amplified by PCR and directly sequenced in each individual. PCR conditions and primer sequences are available on request. Sequencing reactions were performed using Big Dye Terminator v1.1 chemistries (Applied Biosystems, Foster City, CA, USA). Products were then analysed using an ABI 3100 Genetic Analyzer (Applied Biosystems). Sequences were screened for variants using Mutation Surveyor v2.61 software (SoftGenetics, State College, PA, USA).
Results
Table 1 shows the clinical characteristics of the 94 PCGP families with linkage to chromosome 17 markers. Of the families, 83 were Caucasian, 10 were African American, and 1 was Asian American. There were an average of 4.1 confirmed PCa cases per family, and the average age of diagnosis of confirmed PCa cases across families was 63.4 years. In all, 37 families had at least 1 BrCa case and 8 families had at least 1 OvCa case. Five families had both BrCa and OvCa cases. We used the software, Merlin (http://www.sph.umich.edu/csg/abecasis/Merlin/index.html), to calculate the Kong and Cox maximum LOD score on the basis of the exponential model and ‘pairs’ non-parametric allele-sharing statistic at the BRIP1 locus for the families of the 94 patients included in the sequence analysis (Whittemore and Halpern, 1994; Kong and Cox, 1997; Abecasis et al, 2002). The LOD score was 8.26 in these families at the BRIP1 locus.
The 94 individuals sequenced here included 88 men with PCa and 6 women with BrCa. The PCa cases had a median age of diagnosis of 55.5 years, and BrCa cases had a median age of diagnosis of 61.5 years (Table 2). The median pre-diagnosis PSA of PCa cases was 6.15 ng ml−1. The majority of men with PCa were treated surgically, and ∼78% had localised disease at diagnosis.
Overall, no truncating mutations were observed, but a total of 24 SNPs were identified in the sequenced regions of the BRIP1 gene (Table 3). In total, 11 of these SNPs were located in coding regions, including 7 non-synonymous SNPs, 5 SNPs in BRIP1 untranslated regions, and 8 intronic SNPs. Six of the seven non-synonymous SNPs were observed in only one to two individuals. However, the minor allele of S919P was present in 55 of the 94 individuals and had an allele frequency of ∼37%. Overall, none of the individuals was invariant at all 24 SNPs. A total of 59 individuals had at least one missense variant. One individual was heterozygous for three missense variants.
Further analyses were conducted to evaluate the potential function of observed variants. All missense variants were analysed using SIFT (http://sift.jcvi.org/) and PolyPhen (http://genetics.bwh.harvard.edu/pph/) (Ramensky et al, 2002; Ng and Henikoff, 2006), which are computer programmes that use structural and phylogenetic data to predict the functional impact of amino-acid changes. Two SNPs, R264W and R419W, were predicted to be deleterious by both programmes. These SNPs, which were each observed in only one individual, were typed in additional family members. In one family, R264W accounted for two out of three PCa cases and was not present in the unaffected brother of the proband, who was the only unaffected male with DNA available for analysis. In the second family, R419W was present in both the proband and his affected brother; DNA was not available for any other members of this family.
Discussion
The location of BRIP1 near or within the strongest linkage signal identified in a GWS of hereditary PCa families, along with its functional interaction with BRCA1 and its role in BrCa susceptibility, led us to investigate the possibility that BRIP1 is also a PCa susceptibility gene. We screened individuals from families that were highly enriched for both hereditary PCa and chromosome 17 linkage. No obviously deleterious, truncating mutations were detected in the 94 unrelated individuals sequenced from chromosome 17-linked families. However, multiple SNPs, including 7 missense variants, were identified. Although the precise function of these SNPs is unknown, both R264W and R419W were predicted to be deleterious by SIFT and PolyPhen (Ramensky et al, 2002; Ng and Henikoff, 2006).
Truncating BRIP1 mutations have been clearly implicated as cancer susceptibility alleles. Seal et al (2006) identified five different truncating BRIP1 mutations in nine BrCa cases from hereditary BrCa families who had tested negative for BRCA1 and BRCA2 mutations, and reported a relative risk of BrCa of 2.0 for mutation carriers. Additional studies by De Nicolo et al showed that a recombinant protein containing the novel BRIP1 truncating mutation (2992–2995delAAGA) had decreased protein stability and diminished ability to interact with BRCA1. Further experiments using BrCa tissue from a patient carrying the same exon 20 four base-pair deletion confirmed that there was loss of the wild-type allele in tumour cells, consistent with the model of a classical tumour-suppressor gene (De Nicolo et al, 2008). It has also been shown that bi-allelic, truncating mutations in BRIP1 cause Fanconi anaemia complementation group J (Litman et al, 2005).
To date, no study has definitively shown that BRIP1 variants, other than truncating mutations, have a role in susceptibility to any disease. Sigurdson et al (2004) found that women who were homozygous for the serine allele at S919P had a relative risk of developing BrCa of ∼7 before the age of 50 years compared with homozygotes for the proline allele. However, this result was not statistically significant after adjustment for multiple testing. Several subsequent studies have also found no association between S919P genotype and BrCa risk (Garcia-Closas et al, 2006; Vahteristo et al, 2006; Frank et al, 2007). Recently, two BRIP1 tagged SNPs were found to be associated with OvCa risk in case–control samples from the United Kingdom, Denmark, and the United States, but this association was weak and has not been validated (Song et al, 2007).
There is only one report to date that specifically examines the potential contribution of BRIP1 variants to PCa. Kote-Jarai et al (2009) sequenced DNA from the youngest PCa case from each of 192 British multiplex PCa families. They identified a truncating mutation (R798X) in only 1 of the 192 families, and this mutation failed to completely segregate with PCa cases in this family. Sequence analysis identified an additional five non-synonymous variants, including only one variant described in our report (S919P) that was detected in 43% of cases and 46% of controls (N=2081). These authors concluded that truncating BRIP1 mutations may rarely contribute to PCa susceptibility. Although we only sequenced cases from 94 families, compared with 192 in the report by Kote-Jarai, our strategy of selecting chromosome 17-linked families should have increased the likelihood of detecting BRIP1 mutations if they segregated with PCa within these families. The fact that we did not detect any obvious deleterious mutations suggests that BRIP1 mutations are unlikely to contribute to hereditary PCa.
In conclusion, our data suggest that BRIP1 truncating mutations are not common PCa susceptibility alleles and do not account for the chromosome 17 linkage observed in our GWS. The potential role of the coding and non-coding BRIP1 SNPs identified in this report remains unclear, as it is possible that a more common genetic variation contributes to PCa risk. For example, Kote-Jarai et al found significant PCa associations with two SNPs in intron 6 of BRIP1, which are correlated with at least one of the common SNPs identified here (rs4988340; based on the HapMap CEU sample, the pairwise r2=0.89 and 0.78 with rs6505074 and rs8076727, respectively). It is possible that the chromosome 17 linkage observed in multiplex PCGP families is because of mutations in a nearby gene, and the more common BRIP1 variations observed by us and others may have more subtle influences on disease susceptibility. Therefore, follow-up association studies should be considered to assess the possible contribution of these variants to PCa risk.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin-rapid analysis of dense genetic maps using spars gene flow trees. Nat Genet 30: 97–101
Agalliu I, Karlins E, Kwon EM, Iwasaki LM, Diamond A, Ostrander EA, Stanford JL (2007) Rare germline mutations in the BRCA2 gene are associated with early-onset prostate cancer. Br J Cancer 97: 826–831
Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K (2005) The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet 37: 953–957
De Nicolo A, Tancredi M, Lombardi G, Flemma CC, Barbuti S, Di Cristofano C, Sobhian B, Bevilacqua G, Drapkin R, Caligo MA (2008) A novel breast cancer-associated BRIP1 (FANCJ/BACH1) germ-line mutation impairs protein stability and function. Clin Cancer Res 14: 4672–4680
Douglas JA, Levin AM, Zuhlke KA, Ray AM, Johnson GR, Lange EM, Wood DP, Cooney KA (2007) Common variation in the BRCA1 gene and prostate cancer risk. Cancer Epidemiol Biomarkers Prev 16: 1510–1516
Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi R, Hope Q, Osin P, Jackson R, Southgate C, Singh R, Falconer A, Dearnaley DP, Ardern-Jones A, Murkin A, Dowe A, Kelly J, Williams S, Oram R, Stevens M, Teare DM, Ponder BA, Gayther SA, Easton DF, Eeles RA (2003) Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet 72: 1–12
Frank B, Hemminki K, Meindl A, Wappenschmidt B, Sutter C, Kiechle M, Bugert P, Schmutzler RK, Bartram CR, Burwinkel B (2007) BRIP1 (BACH1) variants and familial breast cancer risk: a case-control study. BMC Cancer 7: 83
Garcia-Closas M, Egan KM, Newcomb PA, Brinton LA, Titus-Ernstoff L, Chanock S, Welch R, Lissowska J, Peplonska B, Szeszenia-Dabrowska N, Zatonski W, Bardin-Mikolajczak A, Struewing JP (2006) Polymorphisms in DNA double-strand break repair genes and risk of breast cancer: two population-based studies in USA and Poland, and meta-analyses. Hum Genet 119: 376–388
Gillanders EM, Xu J, Chang BL, Lange EM, Wiklund F, Bailey-Wilson JE, Baffoe-Bonnie A, Jones M, Gildea D, Riedesel E, Albertus J, Isaacs SD, Wiley KE, Mohai CE, Matikainen MP, Tammela TL, Zheng SL, Brown WM, Rokman A, Carpten JD, Meyers DA, Walsh PC, Schleutker J, Gronberg H, Cooney KA, Isaacs WB, Trent JM (2004) Combined genome-wide scan for prostate cancer susceptibility genes. J Natl Cancer Inst 96: 1240–1247
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ (2009) Cancer statistics, 2009. CA Cancer J Clin 59: 225–249
Kirchhoff T, Kauff ND, Mitra N, Nafa K, Huang H, Palmer C, Gulati T, Wadsworth E, Donat S, Robson ME, Ellis NA, Offit K (2004) BRCA mutations and risk of prostate cancer in Ashkenazi Jews. Clin Cancer Res 10: 2918–2921
Kong A, Cox NJ (1997) Alele-sharing models: LOD scores and acurate linkage test. Am J Hum Genet 61: 1179–1188
Kote-Jarai Z, Jugurnauth S, Mulholland S, Leongamornlert DA, Guy M, Edwards S, Tymrakiewitcz M, O’Brien L, Hall A, Wilkinson R, Al Olama AA, Morrison J, Muir K, Neal D, Donovan J, Hamdy F, Easton DF, Eeles R (2009) A recurrent truncating germline mutation in the BRIP1/FANCJ gene and susceptibility to prostate cancer. Br J Cancer 100: 426–430
Kumaraswamy E, Shiekhattar R (2007) Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol 27: 6733–6741
Lange EM, Gillanders EM, Davis CC, Brown WM, Campbell JK, Jones MP, Gildea D, Riedesel E, Albertus J, Freas-Lutz D, Markey C, Giri V, Beebe-Dimmer J, Montie JE, Trent JM, Cooney KA (2003) Genome-wide scan for prostate cancer susceptibility genes using families from the University of Michigan Prostate Cancer Genetics Project finds evidence for linkage on chromosome 17 near BRCA1. Prostate 57: 326–334
Lange EM, Robbins CM, Gillanders EM, Zheng SL, Xu J, Wang Y, White KA, Chang BL, Ho LA, Trent JM, Carpten JD, Isaacs WB, Cooney KA (2007) Fine-mapping the putative chromosome 17q21-22 prostate cancer susceptibility gene to a 10 cM region based on linkage analysis. Hum Genet 121: 49–55
Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB (2005) BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8: 255–265
Ng PC, Henikoff S (2006) Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet 7: 61–80
Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30: 3894–3900
Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, Jack E, Vesprini DJ, Kuperstein G, Abrahamson JL, Fan I, Wong B, Narod SA (2001) Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 68: 700–710
Schaid DJ (2004) The complex genetic epidemiology of prostate cancer. Hum Mol Genet 13 (Spec No 1): R103–R121
Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N (2006) Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 38: 1239–1241
Sigurdson AJ, Hauptmann M, Chatterjee N, Alexander BH, Doody MM, Rutter JL, Struewing JP (2004) Kin-cohort estimates for familial breast cancer risk in relation to variants in DNA base excision repair, BRCA1 interacting and growth factor genes. BMC Cancer 4: 9
Song H, Ramus SJ, Kjaer SK, Hogdall E, DiCioccio RA, Whittemore AS, McGuire V, Hogdall C, Jacobs IJ, Easton DF, Ponder BA, Dunning AM, Gayther SA, Pharoah PD (2007) Tagging single nucleotide polymorphisms in the BRIP1 gene and susceptibility to breast and ovarian cancer. PLoS ONE 2: e268
Thompson D, Easton DF (2002) Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst 94: 1358–1365
Vahteristo P, Yliannala K, Tamminen A, Eerola H, Blomqvist C, Nevanlinna H (2006) BACH1 Ser919Pro variant and breast cancer risk. BMC Cancer 6: 19
Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree memebers. Biometrics 50: 118–127
Xu J, Dimitrov L, Chang BL, Adams TS, Turner AR, Meyers DA, Eeles RA, Easton DF, Foulkes WD, Simard J, Giles GG, Hopper JL, Mahle L, Moller P, Bishop T, Evans C, Edwards S, Meitz J, Bullock S, Hope Q, Hsieh CL, Halpern J, Balise RN, Oakley-Girvan I, Whittemore AS, Ewing CM, Gielzak M, Isaacs SD, Walsh PC, Wiley KE, Isaacs WB, Thibodeau SN, McDonnell SK, Cunningham JM, Zarfas KE, Hebbring S, Schaid DJ, Friedrichsen DM, Deutsch K, Kolb S, Badzioch M, Jarvik GP, Janer M, Hood L, Ostrander EA, Stanford JL, Lange EM, Beebe-Dimmer JL, Mohai CE, Cooney KA, Ikonen T, Baffoe-Bonnie A, Fredriksson H, Matikainen MP, Tammela TL, Bailey-Wilson J, Schleutker J, Maier C, Herkommer K, Hoegel JJ, Vogel W, Paiss T, Wiklund F, Emanuelsson M, Stenman E, Jonsson BA, Gronberg H, Camp NJ, Farnham J, Cannon-Albright LA, Seminara D (2005) A combined genomewide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the international consortium for prostate cancer genetics. Am J Hum Genet 77: 219–229
Yu X, Chini CC, He M, Mer G, Chen J (2003) The BRCT domain is a phospho-protein binding domain. Science 302: 639–642
Zuhlke KA, Madeoy JJ, Beebe-Dimmer J, White KA, Griffin A, Lange EM, Gruber SB, Ostrander EA, Cooney KA (2004) Truncating BRCA1 mutations are uncommon in a cohort of hereditary prostate cancer families with evidence of linkage to 17q markers. Clin Cancer Res 10: 5975–5980
Acknowledgements
We thank the PCGP participants for their role in this study. This study was supported by the University of Michigan Comprehensive Cancer Center and the National Institutes of Health (R01 CA79596) and the National Cancer Institute Specialized Program of Research Excellence in Prostate Cancer (P50 CA69568).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Ray, A., Zuhlke, K., Johnson, G. et al. Absence of truncating BRIP1 mutations in chromosome 17q-linked hereditary prostate cancer families. Br J Cancer 101, 2043–2047 (2009). https://doi.org/10.1038/sj.bjc.6605433
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6605433
Keywords
This article is cited by
-
Mutation analysis of BRIP1 in male breast cancer cases: a population-based study in Central Italy
Breast Cancer Research and Treatment (2011)
