
Abstract
It is generally accepted that the immune system plays an important role in controlling tumour development. However, the interplay between tumour and immune system is complex, as demonstrated by the fact that tumours can successfully establish and develop despite the presence of T cells in tumour. An improved understanding of how tumours evade T-cell surveillance, coupled with technical developments allowing the culture and manipulation of T cells, has driven the exploration of therapeutic strategies based on the adoptive transfer of tumour-specific T cells. The isolation, expansion and re-infusion of large numbers of tumour-specific T cells generated from tumour biopsies has been shown to be feasible. Indeed, impressive clinical responses have been documented in melanoma patients treated with these T cells. These studies and others demonstrate the potential of T cells for the adoptive therapy of cancer. However, the significant technical issues relating to the production of natural tumour-specific T cells suggest that the application of this approach is likely to be limited at the moment. With the advent of retroviral gene transfer technology, it has become possible to efficiently endow T cells with antigen-specific receptors. Using this strategy, it is potentially possible to generate large numbers of tumour reactive T cells rapidly. This review summarises the current gene therapy approaches in relation to the development of adoptive T-cell-based cancer treatments, as these methods now head towards testing in the clinical trial setting.
Similar content being viewed by others
Main
The immune system has developed in order to protect against infection by pathogens and thereby prevent disease. With a greater understanding of immune cell function, there is now an increased awareness that the immune system actually plays a critical role in cancer prevention (Zou, 2005). Delineating this role of the immune system remains a key goal of basic research; however, the implication of these observations is that manipulating and boosting the power of the immune system may prove to be a potent cancer therapy (Murphy et al, 2005).
To this end, immune cells themselves are being increasingly investigated for potential cancer therapies, with a great deal of interest now focused on T cells in particular (Rosenberg et al, 2004). T cells are key effector cells of the adaptive immune system that perform critical activities, which includes target cell lyses, while working in conjunction with other immune cells to orchestrate the immune response through the timed production of cytokines (Shankaran et al, 2001). T cells respond to antigen as a result of the precise interaction of the T-cell receptor (TCR) with antigens presented on target cells. The antigen consists of peptides of varying lengths presented in the binding groove of molecules called the major histocompatibility complex (MHC). Critically, the type of T cell activated is dependent on the MHC presenting the peptide antigen. All nucleated cells express MHC class I proteins that can present antigens to CD8+ expressing T cells, while a more restricted sub-set of cells (generally called antigen-presenting cells (APCs)) express MHC class II, which specifically activate CD4+ T cells. The general defining features of these two sub-sets of T cells are that CD8+ T cells are considered to be the predominant cytotoxic cells (Tc) while CD4+ T cells are thought to play a critical role in refining and optimising T cell and other immune cell responses and so are commonly referred to as helper T cells (Th). It is clear that these separations are simplistic and may not entirely reflect the in vivo functions of the T-cell lineages; however, these distinctions are relevant for a consideration of how T cells can be used for cancer therapy (Figure 1).

Generation of tumour antigen-specific T cells. Different strategies have been employed to endow T cells with the specificity and power to specifically kill tumour. Large numbers of host T cells can be modified to become tumour reactive by transducing them to express. (A) Chimeric immune receptors or (B) tumour-specific T-cell receptors using retroviral technology. (C) Tumour reactive T cells are identified and grown out of a population of tumour infiltrating lymphocytes. These cells are then expanded for use.
Adoptive T-cell therapy: Allogeneic T cells for haemopoietic malignancies
The power of adoptive T-cell therapy has been clearly demonstrated using donor lymphocyte infusions (DLI) for the treatment of a number of haematological malignancies (Kolb et al, 1995). Lymphocytes isolated from an allogeneic donor and given to a patient are thought to respond to tumour through MHC mismatches (either major or minor MHC mismatches) and subsequently eliminate tumour (the graft-vs-tumour effect). However, by the same process, they are also destructive to healthy host tissue. This unwanted side effect, graft-vs-host disease (GvHD), is associated with high rates of morbidity and mortality. This has limited the use of such therapy to a few specialist hospitals, where many strategies have been employed to control such toxicity (Goker et al, 2001). However, T cells are the critical component since their depletion from the DLI abrogates both graft-vs-leukaemia and GvHD effects (Maraninchi et al, 1987).
In order to control GvHD, a novel method is being explored whereby the donor lymphocytes are gene modified to express a suicide gene. The general approach involves the ex vivo transduction of lymphocytes present within the DLI with a retrovirus encoding the suicide gene (e.g. herpes simplex virus thymidine kinase) (Bonini et al, 1997). The DLI is then infused into the patient and, should GvHD develop, the prodrug metabolised by the suicide gene (gancyclovir) is administered to the patient, with the result that the donor T cells are specifically killed thereby controlling toxicity and GvHD (Bonini et al, 1997). Unfortunately, the antitumour response is also diminished as a result of the suicide gene system.
Adoptive T-cell therapy – ‘natural’ T cells
Studies focusing on the use of nonspecific modulators of immune activity such as IL-2 have demonstrated that creditable clinical responses can be achieved in certain tumour types, including melanoma and renal cell cancer (Fyfe et al, 1995; Atkins et al, 1999). These studies have encouraged the development of targeted therapies and, specifically, the clinical testing of antigen-specific T cells for the treatment of melanoma. Antigen-specific T cells isolated directly from tumour biopsies (tumour infiltrating lymphocytes (TILs)) are identified by observing their functional response against melanoma antigens and then subsequently expanded ex vivo by IL-2-driven expansion regimes (Dudley et al, 2002a). While these results have been extensively reviewed (Rosenberg and Dudley, 2004), it is important to consider the salient points of these studies as a platform to consider gene-modified T cells for future clinical studies. A key observation made from these studies showed that both CD4+ and CD8+ T cells are required in order to generate an effective response in vivo (Dudley et al, 2001, 2002b). Furthermore, modulation or elimination of the patient's immune system prior to re-infusion of the expanded T cells was advantageous. A nonmyeloablative preconditioning chemotherapy using cyclophosphamide and fludarabine effectively removed all leukocytes from the patient. The tumour-specific T cells were then re-infused and supported with high-dose IL-2 infusions (Dudley et al, 2002b). Interestingly, clinical responses correlated with T-cell persistence in vivo, suggesting that survival of the T cells was critical to a successful outcome (Meidenbauer et al, 2003). It is not clear whether the beneficial effect of preconditioning was a result of ‘free-space’ created for engraftment through the removal of competing leukocytes or whether the preconditioning had removed important regulatory influences, such as regulatory T cells, thereby allowing the infused T cells to function in a more-friendly immune responsive environment. These studies clearly demonstrated the in vivo effectiveness of antigen-specific T cells and also illustrate that manipulating the environment into which the T cells were being re-infused was also critical. However, it is also clear that generating antigen-specific T cells is highly demanding and requires specialised technical expertise, facilities and equipment. This is due to the fact that antigen-specific T cells represent a very small fraction of the total T-cell population. Subsequently, isolating this small number of cells and expanding them to clinically relevant numbers is an issue of significant proportions. Furthermore, many tumour types do not have a significant TIL population or the tumours themselves are not amenable to surgical removal and/or dissection in order to isolate TILs. Consequently, to date, attempts to use TIL therapy have been effectively restricted to trials in renal cell carcinoma and melanoma. In order to address these issues, gene therapy approaches have been explored in order to facilitate the generation of antigen-specific T cells from peripheral blood.
T cells engineered to express recombinant TCR genes
T cells recognise MHC-peptide conjugates on target cells through the paired α and β chains of the TCR. This pairing confers the antigen specificity of the T cell. One gene therapy approach has involved the molecular cloning of the TCR genes known to be specific for an antigen of choice. These chains are then introduced into T cells usually by means of a retroviral vector. Consequently, expression of the cloned TCRα and TCRβ genes endows the transduced T cell with a functional specificity determined by the pairing of these new genes. In this manner, large numbers of antigen-specific T cells can be generated in a short time period as compared to the longer term culture issues concerning the large-scale expansion of ‘natural T cells’.
There are a number of practical and theoretical issues that are currently being addressed by workers in the field, and recent reviews have provided an in-depth discussion of this specific area (Schumacher, 2002; Willemsen et al, 2003; Stauss et al, 2004). With relevance to this review, there are three principal issues concerning TCR-based gene therapy, which are of broad relevance to oncologists: isolation and expression of the TCR genes, safety and clinical application.
Isolation and expression of suitable TCRα and TCRβ chains
The general methodology involves the isolation of T cells that functionally respond to the target antigen from which the TCR chains are cloned using polymerase chain reaction (PCR)-based methods. The resultant DNA products are sequenced to confirm identity and then placed into retroviral vectors suitable for expression in T cells (Clay et al, 1999). T-cell receptor chains to date have been generated against known antigens with the principal targeted disease being melanoma due to the fact that MHC restricted antigens are numerous and well defined (Rivoltini et al, 2002). Aside from melanoma, there is an increasing diversity of targets being exploited for TCR therapy including MDM-2, a potential target in a wide range of malignancies (Stanislawski et al, 2001) and WT-1 targeting in leukaemia (Xue et al, 2004). However, the rate-limiting step is the identification and isolation of the critical responding T cell to serve as a source to generate the TCR genes with (generally speaking) a great deal of effort generally required to generate antigen-specific T cells. Development of methodologies that will permit the rapid isolation of TCR genes remains a critical area of development, especially when considering that, for widespread use, TCR genes for each HLA type will be required in order to treat every cancer patient (Murphy et al, 2005).
In order to generate a functional T-cell response, both TCRα and TCRβ genes need to be efficiently introduced into the T cell and expressed to levels that will permit sufficient paired complexes to be present on the surface of the transduced T cell. Retroviral gene transfer technologies have been the method of choice since these vectors can efficiently transduce primary T cells (Movassagh et al, 2000). Importantly, current retroviral vectors based on the murine leukaemia virus family require the target cell to be undergoing rapid cell cycling to permit efficient transduction (Miller et al, 1990). With respect to T cells, cell cycling is stimulated by the activation of T cells using strong mitogenic stimuli (with lectins or antibodies), and this may have a profound effect on the subsequent functionality of the transduced T-cell populations. Other vector systems are being investigated (Chinnasamy et al, 2000; Jensen et al, 2000); however, these murine retroviral vectors possess a clinical pedigree (Bonini et al, 2003) and so remain the principal choice for TCR-based approaches. Expression of multiple TCR genes has been achieved through a number of methods, including transduction with multiple retroviruses encoding single genes (Schaft et al, 2003) or single vectors employing internal ribosome entry sites for multiple gene expression (Morgan et al, 2003), although further improvements in vector design are required in order to ensure the high-level expression of multiple genes in retroviral vectors.
Critically, primary human T cells transduced with retroviral vectors encoding TCRs functionally respond against tumour cells expressing the target antigen. These responses include cytokine release, proliferation and cytotoxicity (Gao et al, 2003; Morgan et al, 2003; Schaft et al, 2003), indicating that the overall strategy is highly effective in directing the functional activities of T cells. However, the majority of TCRs cloned to date are MHC class I specific and, consequently, direct the functional activity of CD8+ T cells. Encouragingly, these MHC class I restricted TCR genes also appear to function in CD4+ T cells (Morris et al, 2005), suggesting that a polyclonal T-cell response consisting of both CD4 and CD8 T cells is feasible using a single TCR pairing.
Safety
Tumorigenesis as a result of retroviral insertional mutagenesis has been documented in the case of engineered stem cell therapy for X-linked SCID (Hacein-Bey-Abina et al, 2003). However, at present, there are no examples of clinical manifestation of insertional mutagenesis associated with genetic modification of differentiated T cells (Bonini et al, 2003), suggesting that the same safety issues which are evident in stem cell therapy approaches are a less significant issue in differentiated T-cell therapy (Stauss et al, 2004). However, a specific concern with TCR-based therapy is the fact that the new TCR genes could pair with endogenous TCR genes, with the possible result that T cells with a new and autoimmune specificity could be generated. The probability of this occurring is unknown and the potential danger has to be carefully assessed and balanced against the potential benefit of treating patients with advanced disease. However, more recent studies have focused up engineering the antigen-specific TCRs in order to prevent pairing with endogenous TCR genes (Willemsen et al, 2000).
Clinical application
An important final issue concerning the use of TCR therapy is the fact that dealing with specific TCRs will mean restricting the target patient group to those expressing the correct MHC haplotype, while the reliance upon tumour expression of MHC is central to the therapy. The majority of ongoing studies have focused upon targeting more common MHC haplotypes, however, for widespread application numerous TCR combinations for specific target antigens will have to be developed and validated.
A further complicating factor is that these gene-modified T cells are dependent on the antigen presentation machinery of the tumour. Critically, MHC downregulation is a commonly observed feature of tumours (Garrido et al, 1997). In the absence of a suitable MHC-peptide target on the surface of the tumour, T cells expressing tumour-specific TCR genes would be unlikely to respond against the target. Modulation of MHC expression on tumours is possible; however, in the light of these observations, targeting of T cells to recognise intact cell surface protein antigens using antibody-based technologies, thereby avoiding tumour antigen presentation mechanisms, has been the focus of an increasing number of research groups.
ANTIBODY-TARGETED T CELLS (CHIMERIC IMMUNE RECEPTORS, ENGINEERED T CELLS)
The basis of this field resides in studies performed in the late 1980s, when it was demonstrated that a receptor consisting of a fusion of an antibody domain with the TCRβ chain could direct T-cell hybridomas to respond against the protein antigen targeted by the antibody. This showed that T cells could respond to antigen independent of TCR–MHC interaction (Gross et al, 1989). Given that tumours can frequently lose antigen expression through factors such as the downregulation of MHC expression (Garrido et al, 1997), this makes antibody targeted T cells highly attractive. The subsequent 15 years have seen the rapid development of chimeric immune receptor (CIR) technology through to current testing in ongoing phase I clinical trials. Various recent publications have extensively reviewed this area (Hombach et al, 2002; Thistlethwaite et al, 2005). The aim here is to provide an overview of the current state of antibody targeted T-cell research and to discuss some key scientific areas that are likely to affect upon the immediate translation of this therapy into clinical practice.
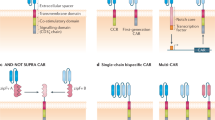
T cells using CIRs to target cellular antigens depend on the introduction of a gene encoding the receptor into the T cell. The essential components of a CIR are an antigen targeting domain fused to an intracellular signalling domain anchored to the surface of the T cell by a transmembrane domain (Figure 2). The antigen binding domain most commonly involves a single chain antibody fragment (scFv) consisting of the antigen recognition components of a monoclonal antibody (Gross et al, 1989; Hawkins et al, 1998), although other protein domains have been successfully used (Mitsuyasu et al, 2000). The scFv maintains the specificity of the original antibody, but carries the advantage of small size suitable for expression as part of a CIR. The predominant requisite for the targeted tumour antigen is cell surface expression. Aside from this, the diversity of antigens targeted to date is extensive with the majority of cancer types represented (Table 1).

The chimeric immune receptor. (A) The T-cell receptor composed of the α and β chains transmits its signal through the CD3 molecule (γ, ɛ, δ and ζ moieties) following interaction with MHC/epitope complex. This differs from the chimeric immune receptor (B) which is composed of an extracellular single chain antibody recognition domain connected to signalling moiety shown in this example as either the CD3 ζ molecule (C) or as a fusion receptor using the CD28 molecule proximal to the ζ moiety. Activation of the chimeric immune receptor can be initiated in the presence of tumour antigen in an MHC independent manner.
The signalling domains used have focused on the key signalling molecules CD3ζ and the γ chain from FcɛRI. A wide number of laboratories have confirmed that CIRs expressed in primary T cells elicit functional responses when cultured with cells expressing the target protein antigen or even purified proteins immobilised on a surface (e.g. on a culture plate) (Haynes et al, 2001). These functional responses (including cytotoxicity and cytokine production) are those thought to be important for antitumour activity. A more recent development has involved the engineering of CIRs to incorporate multiple signalling domains. The first receptor of this type involved a fusion of the CD28 receptor with the CD3ζ moiety (Finney et al, 1998). For full activation of the T cell, multiple signals are required. The TCR/CD3ζ complex generates the antigen-specific response of the T cell (namely cytotoxicity and cytokine production). However, in the absence of associated stimuli (so-called costimulatory signals), the T-cell falters during activation and fails to fully respond to antigen (Brocker and Karjalainen, 1995). Costimulatory signals generated through receptors such as CD28 reinforce the TCR/CD3ζ signal through further cytokine release (including IL-2) and the upregulation of key antiapoptotic gene expression (Krause et al, 1998). Fusion CIRs encoding both CD28 and CD3ζ signalling domains (Figure 2C) would be predicted to function more optimally than a CD3ζ-only receptor since activatory and costimulatory signals would be generated from the same receptor. Indeed, this appears to be the case, with a growing number of reports confirming that T cells expressing CD28-ζ fusion CIRs when stimulated express IL-2 and appear to demonstrate improved antitumour activity in vitro and in vivo (Eshhar et al, 2001; Haynes et al, 2002; Maher et al, 2002). Driven by the relative success of the CD28-based receptors, other known T-cell costimulatory receptors are being tested as CIRs, with the majority appearing to bring significant improvements to the CD3ζ receptor (Finney et al, 2004; Imai et al, 2004).
Once again, there are certain key issues concerning antibody targeted T cells that are likely to be of primary interest to oncologists. These include antigen selection/selection of targeting moiety, safety and clinical application.
Antigen selection/selection of targeting moiety
The key attractive feature of this approach is the generic nature of the targeting construct. Since the target is typically a cell surface expressed tumour associated antigen (TAA), the scFv (or other moiety) is not restricted by HLA expression and, subsequently, a single receptor is broadly applicable to all cancer patients as long as the target antigen is expressed on the tumour. Furthermore, with the development of powerful scFv selection technologies (e.g. phage display (Winter et al, 1994)), scFv can, at least in theory, be rapidly generated against any protein antigen. However, the selection of the target antigen is of critical importance since the expression of the target antigen on normal tissues could result in toxicity (discussed below).
Safety
The issues of retroviral insertional mutagenesis apply to antibody targeted T cells as for TCR gene therapy as discussed previously. However, clinical trials are ongoing, where plasmid-mediated gene transfer has been used to avoid some of the issues with retroviral gene transfer. However, this approach is still likely to suffer from the same potential problems of insertional mutagenesis due to integration of the construct into the genome of the T cell.
The issue of target antigen selection is of major importance. The majority of TAAs are antigens, which are overexpressed on tumours. However, expression of these antigens is not restricted to tumour with antigens typically expressed on normal tissues. For example, carcino-embryoinic antigen (CEA) has been extensively targeted in a range of immunotherapy approaches including antibody targeted T cells (Hombach et al, 1999; Nolan et al, 1999; Gilham et al, 2002). CEA is expressed to high levels on tumour, yet is also expressed to far lower levels on normal gastric mucosal surfaces (Hammarstrom, 1999; Francis et al, 2002). While there has been no evidence of toxicity in CEA vaccination trials (Berinstein, 2002), clinical trials, which are due to start using antibody targeted T cells, will assess whether gut toxicity is observed as a part of the T-cell therapy. In the absence of truly tumour-specific antigens, any therapy targeting tumour antigens is likely to be associated with a level of toxicity. The severity of this toxicity remains to be established and predicting and managing it will be an important component of any trial protocol. Critically, knowledge of the normal tissue distribution of the targeted antigen and the relative level of expression will be important in order to facilitate this process. In the case of CEA, it is envisaged that the high level of expression on tumour cells coupled with the low levels of expression on a restricted distribution of normal cells will mean that autoimmune toxicities should be minimal.
A final point relates to the use of receptor constructs that encode multiple signalling domains. Ligation of CD28-ζ receptor expressing T cells by antigen results in a plethora of signals, which are intended to improve the survival of the engineered T cell. Given that normal cells may express TAAs albeit to low levels, there may be an issue that CD28-ζ receptor T cells could be continuously stimulated and therefore produce longer term unwanted side effects. Once again, the potential toxicity will be balanced by the theoretical improved cancer killing, which may be generated by these improved receptor constructs. Until clinical trials have been carried out against a number of target antigens, it is unclear whether these potential toxicities pose a real or theoretical risk.
Clinical application
Current clinical trials are testing CD3ζ and γ-based CIRs (Lamers et al, 2002) with a trial likely to test CD28-ζ receptors in the near future. While testing the function of engineered T cells in vivo, these trials are also aiming to investigate whether the general approach to gene modification and culture of T cells is feasible.
Future directions
Engineered T-cell therapy is in its infancy, although the approach is now being tested in early-phase clinical trials. These clinical investigations are based on observations, which have shown that engineered T cells (either expressing an engineered TCR or antibody receptor) can respond against their desired antigens in a manner that suggests that these T cells may be effective against cancer in situ. While highly encouraging, these studies also highlight how little is effectively known about engineered T cells. Issues such as how best to culture and gene-modify T cells are still being addressed. The inclusion of cytokines in addition to IL-2 during culture of the T cells is likely to affect the design of clinical protocols in the near future. Trials with TILs suggest that persistence of T cells is important (Robbins et al, 2004), and thus combining the chemotherapy regimes used in TIL trials with engineered T cells may prove to be of major importance. At a molecular level, a basic understanding of how engineered T cells function in response to antigen is also lacking. In addition, the impact of key immune cells that can dampen the immune response (such as regulatory T cells) is not known. There is also a lack of understanding of how gene modification can affect how engineered T cells survive and traffic in the patient's circulation. With these issues in mind, it is likely that combining engineered T cell with preconditioning chemotherapy, IL-2 support and vaccination protocols may all contribute to the in vivo effectiveness of this therapy.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Altenschmidt U, Klundt E, Groner B (1997) Adoptive transfer of in vitro-targeted, activated T lymphocytes results in total tumor regression. J Immunol 159 (11): 5509–5515
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA (1999) High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17: 2105–2116
Berinstein NL (2002) Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol 20: 2197–2207
Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C, Bordignon C (1997) HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 276: 1719–1724
Bonini C, Grez M, Traversari C, Ciceri F, Marktel S, Ferrari G, Dinauer M, Sadat M, Aiuti A, Deola S, Radrizzani M, Hagenbeek A, Apperley J, Ebeling S, Martens A, Kolb HJ, Weber M, Lotti F, Grande A, Weissinger E, Bueren JA, Lamana M, Falkenburg JH, Heemskerk MH, Austin T, Kornblau S, Marini F, Benati C, Magnani Z, Cazzaniga S, Toma S, Gallo-Stampino C, Introna M, Slavin S, Greenberg PD, Bregni M, Mavilio F, Bordignon C (2003) Safety of retroviral gene marking with a truncated NGF receptor. Nat Med 9: 367–369
Brocker T, Karjalainen K (1995) Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med 181: 1653–1659
Chinnasamy D, Chinnasamy N, Enriquez MJ, Otsu M, Morgan RA, Candotti F (2000) Lentiviral-mediated gene transfer into human lymphocytes: role of HIV-1 accessory proteins. Blood 96: 1309–1316
Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI (1999) Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol 163: 507–513
Dudley ME, Wunderlich J, Nishimura MI, Yu D, Yang JC, Topalian SL, Schwartzentruber DJ, Hwu P, Marincola FM, Sherry R, Leitman SF, Rosenberg SA (2001) Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother 24: 363–373
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA (2002a) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298: 850–854
Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA, Rogers-Freezer L, Morton KE, Nahvi A, Mavroukakis SA, White DE, Rosenberg SA (2002b) A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother 25: 243–251
Eshhar Z, Waks T, Bendavid A, Schindler DG (2001) Functional expression of chimeric receptor genes in human T cells. J Immunol Methods 248: 67–76
Finney HM, Akbar AN, Lawson AD (2004) Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol 172: 104–113
Finney HM, Lawson AD, Bebbington CR, Weir AN (1998) Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol 161: 2791–2797
Francis RJ, Sharma SK, Springer C, Green AJ, Hope-Stone LD, Sena L, Martin J, Adamson KL, Robbins A, Gumbrell L, O’Malley D, Tsiompanou E, Shahbakhti H, Webley S, Hochhauser D, Hilson AJ, Blakey D, Begent RH (2002) A phase I trial of antibody directed enzyme prodrug therapy (ADEPT) in patients with advanced colorectal carcinoma or other CEA producing tumours. Br J Cancer 87: 600–607
Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC (1995) Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol 13: 688–696
Gao L, Xue SA, Hasserjian R, Cotter F, Kaeda J, Goldman JM, Dazzi F, Stauss HJ (2003) Human cytotoxic T lymphocytes specific for Wilms’ tumor antigen-1 inhibit engraftment of leukemia-initiating stem cells in non-obese diabetic-severe combined immunodeficient recipients. Transplantation 75: 1429–1436
Garrido F, Ruiz-Cabello F, Cabrera T, Perez-Villar JJ, Lopez-Botet M, Duggan-Keen M, Stern PL (1997) Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today 18: 89–95
Gilham DE, O’Neil A, Hughes C, Guest RD, Kirillova N, Lehane M, Hawkins RE (2002) Primary polyclonal human T lymphocytes targeted to carcino-embryonic antigens and neural cell adhesion molecule tumor antigens by CD3zeta-based chimeric immune receptors. J Immunother 25: 139–151
Goker H, Haznedaroglu IC, Chao NJ (2001) Acute graft-vs-host disease: pathobiology and management. Exp Hematol 29: 259–277
Gross G, Waks T, Eshhar Z (1989) Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 86: 10024–10028
Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, Fischer A (2003) A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 348: 255–256
Hammarstrom S (1999) The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 9: 67–81
Hawkins RE, Whittington HA, Watkins SJ, Gilham DE (1998) Antibodies: from genes to targeted cancer gene therapy. Gene Therapy 5: 1581–1583
Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ, Darcy PK (2001) Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J Immunol 166: 182–187
Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, Kershaw MH, Smyth MJ, Darcy PK (2002) Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol 169: 5780–5786
Hombach A, Heuser C, Abken H (2002) The recombinant T cell receptor strategy: insights into structure and function of recombinant immunoreceptors on the way towards an optimal receptor design for cellular immunotherapy. Curr Gene Ther 2: 211–226
Hombach A, Koch D, Sircar R, Heuser C, Diehl V, Kruis W, Pohl C, Abken H (1999) A chimeric receptor that selectively targets membrane-bound carcinoembryonic antigen (mCEA) in the presence of soluble CEA. Gene Therapy 6: 300–304
Hombach A, Sent D, Schneider C, Heuser C, Koch D, Pohl C, Seliger B, Abken H (2001) T cells engrafted with a recombinant anti-CD30 receptor target autologous CD30(+) cutaneous lymphoma cells. Gene Therapy 8 (11): 891–895
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D (2004) Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18: 676–684
Jensen MC, Clarke P, Tan G, Wright C, Chung-Chang W, Clark TN, Zhang F, Slovak ML, Wu AM, Forman SJ, Raubitschek A (2000) Human T lymphocyte genetic modification with naked DNA. Mol Ther 1: 49–55
Jensen MC, Cooper LJ, Wu AM, Forman SJ, Raubitschek A (2003) Engineered CD20-specific primary human cytotoxic T lymphocytes for targeting B-cell malignancy. Cytotherapy 5 (2): 131–138
Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, Ljungman P, Ferrant A, Verdonck L, Niederwieser D, van Rhee F, Mittermueller J, de Witte T, Holler E, Ansari H (1995) Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. European Group for Blood and Marrow Transplantation Working Party Chronic Leukemia. Blood 86: 2041–2050
Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M (1998) Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med 188: 619–626
Lamers CH, Willemsen RA, Luider BA, Debets R, Bolhuis RL (2002) Protocol for gene transduction and expansion of human T lymphocytes for clinical immunogene therapy of cancer. Cancer Gene Ther 9: 613–623
Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M (2002) Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol 20: 70–75
Maraninchi D, Gluckman E, Blaise D, Guyotat D, Rio B, Pico JL, Leblond V, Michallet M, Dreyfus F, Ifrah N, Bordigoni A (1987) Impact of T-cell depletion on outcome of allogeneic bone-marrow transplantation for standard-risk leukaemias. Lancet 2: 175–178
Meidenbauer N, Marienhagen J, Laumer M, Vogl S, Heymann J, Andreesen R, Mackensen A (2003) Survival and tumor localization of adoptively transferred Melan-A-specific T cells in melanoma patients. J Immunol 170: 2161–2169
Miller DG, Adam MA, Miller AD (1990) Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol 10: 4239–4242
Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, Bakker A, Roberts MR, June CH, Jalali S, Lin AA, Pennathur-Das R, Hege KM (2000) Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 96: 785–793
Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, Wunderlich JR, Hughes MS, Restifo NP, Rosenberg SA (2003) High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol 171: 3287–3295
Morris EC, Tsallios A, Bendle GM, Xue SA, Stauss HJ (2005) A critical role of T cell antigen receptor-transduced MHC class I-restricted helper T cells in tumor protection. Proc Natl Acad Sci USA 102: 7934–7939
Movassagh M, Boyer O, Burland MC, Leclercq V, Klatzmann D, Lemoine FM (2000) Retrovirus-mediated gene transfer into T cells: 95% transduction efficiency without further in vitro selection. Hum Gene Ther 11: 1189–1200
Murphy A, Westwood JA, Teng MW, Moeller M, Darcy PK, Kershaw MH (2005) Gene modification strategies to induce tumor immunity. Immunity 22: 403–414
Nolan KF, Yun CO, Akamatsu Y, Murphy JC, Leung SO, Beecham EJ, Junghans RP (1999) Bypassing immunization: optimized design of ‘designer T cells’ against carcinoembryonic antigen (CEA)-expressing tumors, and lack of suppression by soluble CEA. Clin Cancer Res 5: 3928–3941
Rivoltini L, Carrabba M, Huber V, Castelli C, Novellino L, Dalerba P, Mortarini R, Arancia G, Anichini A, Fais S, Parmiani G (2002) Immunity to cancer: attack and escape in T lymphocyte–tumor cell interaction. Immunol Rev 188: 97–113
Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell Jr DJ, Rosenberg SA (2004) Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol 173: 7125–7130
Rosenberg SA, Dudley ME (2004) Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA 101 (Suppl 2): 14639–14645
Rosenberg SA, Yang JC, Restifo NP (2004) Cancer immunotherapy: moving beyond current vaccines. Nat Med 10: 909–915
Schaft N, Willemsen RA, de Vries J, Lankiewicz B, Essers BW, Gratama JW, Figdor CG, Bolhuis RL, Debets R, Adema GJ (2003) Peptide fine specificity of anti-glycoprotein 100 CTL is preserved following transfer of engineered TCR alpha beta genes into primary human T lymphocytes. J Immunol 170: 2186–2194
Schumacher TN (2002) T-cell-receptor gene therapy. Nat Rev Immunol 2: 512–519
Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD (2001) IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410: 1107–1111
Stanislawski T, Voss RH, Lotz C, Sadovnikova E, Willemsen RA, Kuball J, Ruppert T, Bolhuis RL, Melief CJ, Huber C, Stauss HJ, Theobald M (2001) Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol 2: 962–970
Stauss HJ, Xue S, Gillmore R, Gao L, Bendle G, Holler A, Downs AM, Morris E (2004) Exploiting alloreactivity for tumour immunotherapy. Vox Sang 87 (Suppl 2): 227–229
Thistlethwaite F, Mansoor W, Gilham DE, Hawkins RE (2005) Engineering T-cells with antibody-based chimeric receptors for effective cancer therapy. Curr Opin Mol Ther 7: 48–55
Walker RE, Bechtel CM, Natarajan V, Beseler M, Hege KM, Metcalf JA, Stevens R, Hazen A, Blaese RM, Chen CC, Leitman SF, Palensky J, Wittes J, Davey Jr RT, Falloon J, Polis MA, Kovacs JA, Broad DF, Levine BL, Roberts MR, Masur H, Lane HC (2000) Long-term in vivo survival of receptormodified syngeneic T cells in patients with human immunodeficiency virus infection. Blood 96 (2): 467–474
Weijtens ME, Hart EH, Bolhuis RL (2000) Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Therapy 7 (1): 35–42
Willemsen RA, Debets R, Chames P, Bolhuis RL (2003) Genetic engineering of T cell specificity for immunotherapy of cancer. Hum Immunol 64: 56–68
Willemsen RA, Weijtens ME, Ronteltap C, Eshhar Z, Gratama JW, Chames P, Bolhuis RL (2000) Grafting primary human T lymphocytes with cancer-specific chimeric single chain and two chain TCR. Gene Therapy 7: 1369–1377
Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR (1994) Making antibodies by phage display technology. Annu Rev Immunol 12: 433–455
Xue S, Gao L, Gillmore R, Bendle G, Holler A, Downs AM, Tsallios A, Ramirez F, Ghani Y, Hart D, Alcock S, Tranter A, Stauss HJ, Morris E (2004) WT1-targeted immunotherapy of leukaemia. Blood Cells Mol Dis 33: 288–290
Zou W (2005) Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5: 263–274
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Mansoor, W., Gilham, D., Thistlethwaite, F. et al. Engineering T cells for cancer therapy. Br J Cancer 93, 1085–1091 (2005). https://doi.org/10.1038/sj.bjc.6602839
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6602839
Keywords
This article is cited by
-
Injectable cryogel-based whole-cell cancer vaccines
Nature Communications (2015)
-
Retargeting NK‐92 for anti‐melanoma activity by a TCR‐like single‐domain antibody
Immunology & Cell Biology (2013)
-
Redirected Antitumor Activity of Primary Human Lymphocytes Transduced With a Fully Human Anti-mesothelin Chimeric Receptor
Molecular Therapy (2012)
-
Antitumor activity of dual-specific T cells and influenza virus
Cancer Gene Therapy (2007)
