
Abstract
ABC transporter proteins may protect haematopoietic progenitor cells from chemotherapy-induced toxicity. By using an in vitro colony-forming assay, we found that bone marrow of Mdr1ab, Mrp1, Mdr1ab/Mrp1 knockout (KO) mice was two-, five- to 10- and 25-fold, respectively, more sensitive to vincristine than wild-type mice bone marrow. To study the impact of ABC transporters on in vivo bone marrow sensitivity without the added complication of altered pharmacokinetics, we created chimeras of wild-type mice transplanted with bone marrow from wild-type, Mrp1, Mdr1ab or Mdr1ab/Mrp1 KO donor mice. Following a single bolus injection of vincristine, the chimeras transplanted with wild-type or Mdr1ab KO marrow cells showed no reductions in WBC. A significant reduction was observed in Mrp1 KO chimeras, but the most pronounced effect was observed in mice receiving bone marrow from Mdr1ab/Mrp1 KO mice. A pharmacokinetic analysis in wild-type and KO mice showed that the absence of P-gp reduced the body clearance of vincristine, but that no further reduction occurred when Mrp1 was also absent. However, the tissue accumulation of vincristine in tissues of these Mdr1ab/Mrp1 KO mice was further increased. This study demonstrates that the presence of multiple drug transporters protects the bone marrow, and probably other tissues as well, against chemotherapeutic insults.
Similar content being viewed by others


Main
ABC transporters such as P-glycoprotein (P-gp) provide efficient protection against xenobiotics. As demonstrated by using knockout (KO) mouse models, P-gp and Breast cancer resistance protein (Bcrp1) limit the uptake from the gastrointestinal tract of substances, such as drugs (Sparreboom et al, 1997; Jonker et al, 2000) or potentially toxic dietary substances (Jonker et al, 2002). Similarly, P-gp in the blood–brain barrier limits the entry of substances into the brain (Schinkel et al, 1994, 1996). Multidrug resistance-associated protein (Mrp1) KO mice were hypersensitive to etoposide (Lorico et al, 1997; Wijnholds et al, 1997). Mrp1 protects the oropharyngeal mucosal layer and the testicular tubules (Wijnholds et al, 1998) and plays a role in the blood–cerebrospinal fluid barrier (Rao et al, 1999; Wijnholds et al, 2000).
Myelotoxicity is a common complication when treating cancer patients with chemotherapeutic drugs. Already in 1991, Chaudhary and Roninson (1991) showed that P-gp was present in haematopoietic progenitor cells, suggesting that this transporter might also be involved in the protection of bone marrow stem cells. The role of drug transporters in bone marrow-derived cells under in vitro conditions was further addressed in several studies in vitro. Mdr1ab P-gp contributed to the extrusion of the fluorophore rhodamine 123 from haematopoietic progenitor cells (Schinkel et al, 1997). Moreover, it was recently shown that Bcrp1 is the transport protein responsible for the extrusion of the marker dye Hoechst 33342 (Zhou et al, 2001; Kim et al, 2002; Scharenberg et al, 2002). Flow cytometry cell sorting of the fraction of Hoechst 33342 dull cells resulted in a highly enriched fraction of primitive haematopoietic progenitor cells. Subsequent RT–PCR analyses of RNA from these so-called side-population (SP) cells revealed the presence of Mrp1, Mrp3 and Mrp4 but not Mrp2 (Zhou et al, 2001).
The role of these drug transporters in vivo is currently under investigation. Bcrp1 appears to protect the bone marrow from mitoxantrone-induced toxicity (Zhou et al, 2002). Johnson et al (2001) showed that mice with compound disruptions of the Mdr1a, Mdr1b and the Mrp1 genes (Mdr1ab/Mrp1 triple KO mice) were about 128-fold more sensitive for i.p. vincristine. Their results suggested that the bone marrow might be involved in the toxicity profile, but it remained unclear whether the toxicity was directly due to the absence of these drug transporters in the haematopoietic progenitor cells or whether it was also, or even merely, a consequence of a reduced drug clearance in these Mdr1ab/Mrp1 triple KO mice. The pharmacokinetics of vincristine has not been documented, but it is well established that the absence of P-gp reduces the clearance of substrate drugs, such as vinblastine (van Asperen et al, 1996).
In the present study, we have investigated the role of P-gp and Mrp1 in the protection of the bone marrow in vitro and in vivo. We have used a bone marrow transplantation model to avoid complications in data interpretation due to differences in drug clearance. Wild-type mice receiving whole-body irradiation at a dose that was lethal to the bone marrow were transplanted with bone marrow from donor mice deficient in Mdr1ab and/or Mrp1 genes. Animals transplanted with wild-type bone marrow were used as control group. After their full recovery, the mice were exposed to the anticancer drug vincristine and the bone marrow toxicity was determined by serial analyses of haematology parameters. In parallel experiments, the pharmacokinetics and toxicology of vincristine in the wild-type and Mdr1ab/Mrp1 triple KO mice were established using a selective high-performance liquid chromatographic (HPLC) assay.
Materials and methods
Animals
Male wild-type, Mrp1 knockout (Wijnholds et al, 1997), Mdr1ab double knockout (Mdr1ab DKO; animals with compound deletion of Mdr1a and Mdr1b genes) (Schinkel et al, 1997) and Mdr1ab/Mrp1 triple knockout (Mdr1ab/Mrp1 TKO) were used. The latter were obtained by crossbreeding of Mrp1 KO and Mdr1ab DKO mice. All mouse strains were backcrossed for at least seven generations to obtain a more than 99% homogeneous FVB background. The animals were given food (Hope Farms BV, Woerden, The Netherlands) and acidified water ad libitum. They were handled according to the institutional guidelines, which are based on Dutch law and conform the standards required by the UKCCCR guidelines (Workman et al, 1998). The animal experiment committee of the Institute approved the experiments described in this paper.
Determination of the maximum tolerated dose (MTD)
Vincristine (Pharmachemie, Haarlem, The Netherlands; 1 mg ml−1) was diluted in saline and administered i.v. to wild-type and Mdr1ab/Mrp1 TKO mice, aged 10–14 weeks, at dose levels ranging between 0.125 and 4 mg kg−1. Animals were monitored daily and killed when they lost more than 20% of their initial body weight. The MTD was defined as one dose step below the dose where more than one animal in that group had to be killed. Necropsies were performed in wild-type and Mdr1ab/Mrp1 TKO mice receiving vincristine at or near the MTD and killed 2 days later.
Pharmacokinetics
The pharmacokinetic behaviour of vincristine in wild-type, Mdr1ab KO and Mdr1ab/Mrp1 TKO mice, 10–14 weeks of age, was established at a dose of 1 mg kg−1. Animals were killed at 5, 15, 30 min, 1, 2 and 4 h after drug administration for collection of plasma. At 1 and 4 h, a range of tissues was also collected. Vincristine levels in plasma and tissues were analysed by HPLC developed by us previously (Boven et al, 1999). Plasma AUC0–4 h (area under the curve) values were calculated by the linear trapezoidal rule using standard equations (van Asperen et al, 1996) and the clearance was calculated as Dose/AUC0–4 h. Statistical tests were performed with SPSS v11.0 (SPSS Inc., Chicago, IL, USA).
In vitro bone marrow toxicity
The toxicity of vincristine in haematopoietic progenitor cells was tested by an in vitro colony forming unit (CFU) assay. Mouse bone marrow progenitor cells were obtained from the femurs of FVB mice flushed with Dulbecco's phosphate-buffered saline solution. After centrifugation (10 min, 200 g, ambient temperature), the cell pellet was resuspended in Iscoves medium (MDM) (StemCell Technologies Inc., Vancouver, BC, Canada) with 2% (v v−1) fetal calf serum (FCS). Nucleated bone marrow cells were seeded in Methocult GF M3534 at a density of 2.5 × 105 cells ml−1 and vincristine was added at concentrations ranging from 2.5 to 100 ng ml−1. Aliquots of 1 ml were plated in duplicate in uncoated six-wells culture plates (Greiner, Alphen a/d Rijn, The Netherlands) and incubated for 5–6 days at 37oC in 5% CO2 in humidified air. CFUs were scored by phase-contrast light microscopy. We have used the concentration that reduces the number of colonies by more than 90% relative to untreated controls (IC90) instead of IC50 because the decreasing size of the colonies at higher drug concentrations makes accurate scoring of their numbers difficult. Hardly any colonies were present at the IC90 making this parameter easier to determine.
In vivo bone marrow toxicity
Wild-type mice of about 5–6 weeks of age received whole-body irradiation of 6.8 Gy (HF320 Radiobiology Constant Potential X-ray Unit, Pantak, East Haven, CT, USA). The next morning each mouse was i.v. injected with 1.5 to 3 × 106 nucleated bone marrow cells from donor mice of wild-type, mrp1 KO, Mdr1ab DKO or Mdr1ab/Mrp1 TKO genotype. After a recovery period of 6 weeks, the mice received an i.v. bolus injection of 2 mg kg−1 of vincristine via the tail vein. To minimise the effects of mild dehydration occurring at this dose level of vincristine, mice were supported by daily i.p. administrations of 1 ml of saline: dextrose 5% (1 : 1; v/v) for 3 days. At days 0 (before vincristine), 2, 4, 7 and 12, peripheral blood was sampled from the tail and haematologic parameters (WBC and Haemoglobin (Hb)) were determined on a Cell Dyn 1200 analyzer (Abbott Laboratories, Santa Clara, CA, USA). Experiments were performed on six different occasions. At some of these occasions, the mice receiving bone marrow of TKO mice were also challenged with vincristine at lower dose levels (e.g. 0.5 or 1 mg kg−1). Bone marrow was obtained at the end of the experiments from a number of randomly selected mice and used to determine the in vitro sensitivity of the grafted bone marrow using the in vitro bone marrow toxicity test.
PCR analyses
As a random test, the genotype of the engrafted bone marrow was verified by PCR analyses in about 50% of all animals. DNA was prepared from whole-blood samples of recipient animals using the DNeasy kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer's protocol. Disruption of genes in the KO mice strains was achieved by replacement of relevant genomic fragments by a selection gene, for example, hygromycin in case of Mdr1a (4). As a result, PCR verification of the Mdr1a KO genotype was verified by the presence and absence of bands for hygromycin and Mdr1a, respectively.
Results
We first examined the relative sensitivities of bone marrow derived from wild type, Mrp1 KO, Mdr1ab DKO and Mdr1ab/Mrp1 TKO mice using an in vitro CFU assay. Cell kill occurred in a dose-dependent manner. The bone marrow of wild-type animals was most resistant to vincristine (Table 1). Mdr1ab DKO bone marrow cells were about two-fold more sensitive, whereas Mrp1 KO cells were about five to 10-fold more sensitive. The absence of both drug transporters in cells from the Mdr1ab/Mrp1 TKO mice resulted in an about 25-fold higher susceptibility towards vincristine. These experiments were repeated several times and although the absolute IC90 values varied with different batches of culture medium, the relative differences in IC90 between the bone marrow of the various genotypes were consistent throughout these experiments.
We next investigated the MTD of vincristine in our mouse strains (Table 2). Based on our previous experience, we selected a few dose levels in wild-type animals (1, 2 and 4 mg kg−1). The MTD in wild-type mice was 2 mg kg−1, where the mice experienced mild but clear signs of toxicity, including pilo-erection, and a median weight loss of about 12% of the initial value achieved around day 2, followed by weight gain thereafter. Only one wild-type mouse receiving 2 mg kg−1 experienced body weight loss in excess of 20%; however, this nadir was reached at 7 days after drug administration at a time that surviving animals were already regaining weight. It was not clear whether this event was due to vincristine toxicity or to other causes. A dose level of 4 mg kg−1 was clearly above the MTD, since five out of five animals experienced more than 20% body weight loss.
The MTD in Mdr1ab/Mrp1 TKO mice was clearly lower at 0.5 mg kg−1, but similar macroscopic signs of toxicity were observed. At a dose of 1 mg kg−1, two out of four animals had to be killed (at days 4 and 5) because their body weight dropped by more than 20%, whereas the two others survived. These results indicate that there is a moderate increase in vincristine toxicity in animals lacking both these drug-transporting proteins.
Toxic effects of vincristine in wild-type and Mdr1ab/Mrp1 TKO mice were assessed by full necropsy of the animals 2 days after they received vincristine at their respective MTD. In Mdr1ab/Mrp1 TKO mice receiving 0.5 mg kg−1 toxicities manifested by arrested mitoses were found in the intestines, skin, bone marrow, the adrenal medulla and in the bases of the incisors. No evidence for central nervous toxicity or liver toxicity was observed. In wild-type mice receiving a dose of 2 mg kg−1, the incisors were affected and a moderate depletion of the bone marrow was seen, but no signs of toxicity were found in any of the other tissues, except for some mild vacuolisation of spinal ganglion neurons.
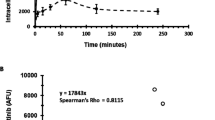
We further investigated the pharmacokinetics of vincristine given at a dose of 1 mg kg−1 to wild-type, Mdr1ab DKO and Mdr1ab/Mrp1 TKO mice by sampling of blood and tissues for up to 4 h after drug administration. Although this dose level was above the MTD in Mdr1ab/Mrp1 TKO mice, it was selected because toxic effects are relatively mild during the first 4 h and because of the detection limit of the assay (5 ng ml−1 for a 200 μl plasma sample). In line with the toxicity results there was only a moderate reduction in the plasma clearance of vincristine (Figure 1), and this appears to be due mainly to the absence of P-gp, since the plasma concentration–time curves of Mdr1ab DKO and Mdr1ab/Mrp1 TKO mice are overlapping. The plasma clearance in wild-type mice was 16.2±1.0 μg ml−1 h and was significantly (P<0.01) reduced to 12.0±0.6 and 12.1±0.5 μg ml−1 h in Mdr1ab DKO and Mdr1ab/Mrp1 TKO mice, respectively. Similar as in plasma, the vincristine levels observed in tissues of Mdr1ab DKO were significantly higher than in wild-type mice (Figure 2). Interestingly, however, the levels of vincristine in several tissues of Mdr1ab/Mrp1 TKO were again significantly higher than those in Mdr1ab DKO, despite equivalent plasma levels, suggesting that Mrp1 offers additional protection against vincristine accumulation in these tissues. The vincristine levels in the brain were below the limit of detection in all animals.

Plasma concentration–time profiles of vincristine. Mice received 1 mg kg−1 of vincristine by i.v. injection. Each time point represents at least four animals. The AUC values were calculated by the linear trapezoidal rule and were 61.8±3.8, 83.1±4.2 and 82.4±3.4 ng ml−1 h for wild type, Mdr1ab DKO and Mdr1ab/Mrp1 TKO, respectively. The latter two were significantly higher (P<0.01) than the values of wild-type mice, but not significantly different from each other.

Tissue accumulation of vincristine. Mice receiving 1 mg kg−1 were killed at 1 and 4 h after drug administration. Drug levels were determined by HPLC. Each bar represents at least four animals and the error bar depicts the s.e. Statistical analyses were performed by ANOVA using Bonferroni post hoc test for multiple comparisons. * P<0.05 relative to wild-type; ** P<0.05 relative to Mdr1ab DKO, otherwise not significant.
To avoid the complicating effects of these transporters on the pharmacokinetics of vincristine, we investigated the consequences of the absence of the various transporters on the bone marrow in vivo using animals with transplanted bone marrows of different genotype. By using this approach we ensured that the only difference in these mice was in the genotype of their bone marrow. Wild-type mice, which tolerate the highest dose of vincristine, received whole-body irradiation at a dose of 6.8 Gy. All mice that were not supplemented with donor bone marrow died within 10 days after irradiation when the WBC and the Hb concentration had dropped to extremely low values. With a few exceptions, the mice that did receive bone marrow of donor mice remained in good condition. These results show that irradiation induced bone marrow ablation had occurred and was the primary cause of death. Wild-type animals received donor bone marrow of either wild-type, Mrp1 KO, Mdr1ab DKO or Mdr1ab/Mrp1 TKO mice, and were allowed to recover for an additional 6 weeks before receiving one cycle of vincristine chemotherapy by i.v. administration. Most animals were treated with the MTD of vincristine for wild-type mice (2 mg kg−1).
Both in wild type and in Mdr1ab DKO chimeras, there was no reduction in WBC (Figure 3). On the contrary, the levels increased after day 2, which may be due to chemotherapy-induced recruitment of cells from the bone marrow or due to the procedure of blood sampling from the tail vein; a procedure that may cause a mild inflammatory response. The WBC counts in animals receiving Mrp1 KO bone marrow had dropped significantly at day 4, but showed a steep rebound by day 7. The most pronounced effect on WBC was observed in mice receiving Mdr1ab/Mrp1 TKO donor marrow. The nadir in these mice occurred at day 4 followed by a gradual recovery to baseline values within about 12 days. The nadir in WBC in Mdr1ab/Mrp1 chimeras was clearly dose dependent (Figure 3: right panel).

In vivo bone marrow toxicity induced by vincristine: effects on WBC. Lethally irradiated wild-type mice, which had received donor marrow from wild-type (n=38), Mrp1 KO (n=33), Mdr1ab DKO (N=18) and Mdr1ab/Mrp1 TKO (n=38) mice, were challenged with 2 mg kg−1 of vincristine administered by i.v. injection (upper panel). The course of the WBC in blood is depicted as percentage of the initial value at day 0 (arbitrarily set at 100%). * P<0.05 relative to wild type, otherwise not significant. The lower panel depicts the results in Mdr1ab/Mrp1 TKO donor marrow grafted mice receiving 0.5 mg (n=15), 1 mg (n=20) or 2 mg (n=21) of vincristine per kilogram and show the dose-dependent response on WBC.
The Hb concentrations in wild-type and Mdr1ab KO chimeras increased slightly by day 2, which may be due to the mild dehydration as a consequence of the toxicity occurring at this dose of vincristine (Figure 4). In Mdr1ab/Mrp1 TKO chimeras receiving a lower dose (0.5 or 1 mg kg−1) of vincristine, this effect was not observed (not shown). By day 12, the Hb level in Mdr1ab/Mrp1 TKO chimeras was significantly lower than observed in the other cohorts (P<0.001). Overall, however, the effect on Hb was less severe than the effect on the WBC counts, which is probably due to the longer half-life of mature red blood cells compared to peripheral WBC.

In vivo bone marrow toxicity induced by vincristine: effects on Hb levels. Lethally irradiated wild-type mice received donor marrow from wild-type, Mrp1 KO, Mdr1ab DKO and Mdr1ab/Mrp1 (TKO) mice were challenged by i.v. administration of 2 mg of vincristine per kilogram (left panel). The course of the haemoglobin concentration is depicted as percentage of the initial value at day 0 (arbitrarily set at 100%). * P<0.05 relative to wild type; otherwise not significant.
We performed a random check whether the repopulation of the bone marrow by donor mice was complete or only partial by harvesting of bone marrow of animals at the end of the study, which was and in the in vitro CFU assay. In all cases, bone marrow derived from Mdr1ab/Mrp1 chimeras showed the same high sensitivity to vincristine as bone marrow obtained from Mdr1ab/Mrp1 TKO mice used as control. In addition, PCR analyses of whole-blood samples from the mice at the end of the experiment were used to verify the genotype of the donor bone marrow. Although the results for the hygromycin gene (selection marker for the Mdr1a KO allele) were consistent in all cases, we observed a weak signal for Mdr1a in mice that received bone marrow of Mdr1ab DKO or Mdr1ab/Mrp1 TKO donor mice. This is probably due to the high sensitivity of the PCR assay, which detects small quantities of host genomic DNA in the blood. It was estimated that up to 5% of circulating nucleated cells were of wild-type origin.
Discussion
This study using an in vivo model shows that the ABC-transporters P-gp and Mrp1 protect haematopoietic progenitor cells in the bone marrow from vincristine toxicity. The most significant effects in WBC and Hb values were observed in mice grafted with bone marrow from Mdr1ab/Mrp1 TKO mice. Mrp1 chimeras showed intermediate signs of haematotoxicity, whereas no toxicity was observed in animals that received bone marrow from Mdr1ab DKO or wild-type donor mice. Thus, in the case of vincristine, the two transporters appear to function in concert in bone marrow progenitor cells and the alternate transporter can partly or completely compensate the loss of function of the other. Overall, the lethality in Mdr1ab/Mrp1 TKO chimeras after challenging with vincristine was a few percent and not higher than in animals bearing Mdr1ab DKO, Mrp1 KO or wild-type bone marrow. This shows that the increased haematotoxicity after single high-dose vincristine treatment was not a dose-limiting event per se.
A pharmacokinetic analysis of vincristine showed that, relative to wild-type mice, the plasma clearance of vincristine was reduced in Mdr1ab DKO mice but not further reduced in Mdr1ab/Mrp1 TKO mice. This result is in line with the location of P-gp in apical membranes of excretory organs (liver, intestines, kidneys) (Thiebaut et al, 1987), where it is involved in detoxification by extrusion of substrates from the body, whereas Mrp1 is mainly located in basolateral membranes and has relatively little direct effect on drug excretion from the body (Borst et al, 1999; Hipfner et al, 1999). However, this study shows that the accumulation of vincristine in tissues from Mdr1ab/Mrp1 TKO mice was clearly enhanced relative to Mdr1ab DKO mice showing that Mrp1 offers protection to tissues. A previous study in Mrp1 KO mice did not find any effects on tissue distribution of etoposide (Wijnholds et al, 1997). This discrepancy may be (partly) due to the presence of P-gp in these mice, which may be such a dominant factor that it conceals the effects of a loss of Mrp1. However, it is also possible that the analytical methodology (determination of total radioactivity after administration of radiolabelled drug) was not suited to find these differences, because it cannot discern unchanged substrate drug from (radiolabelled) metabolites and/or degradation products. We have used a selective HPLC to determine unchanged vincristine levels. The higher drug levels in tissues correlates with previous reports of local organ toxicities when Mrp1 KO mice were challenged with chemotherapeutics (Wijnholds et al, 1998). It thus seems likely that P-gp located at the apical side and Mrp1 at the basolateral side of epithelia cooperate in limiting the accumulation of compounds that are substrates of both transporters, such as vincristine, whereas they are less efficient for compounds that are substrate of only one of them. Moreover, their concerted activity has also been shown in cells expressing these transporters in a nonpolarised fashion. By using immortalised fibroblast cell lines, it was found that both Mdr1ab and Mrp1 are implicated in innate resistance to cytotoxic substrates (Allen et al, 2000; Lin et al, 2002), but the strongest effects were found in cells lacking both transporters. Our findings of a markedly enhanced toxicity by vincristine in the in vitro bone marrow toxicity assays are in line with these results.
The dose-limiting toxicity of vincristine in Mdr1ab/Mrp1 TKO mice appears to be related to the effects on the gastrointestinal tract. We found that the Mdr1ab/Mrp1 TKO mice were only about four-fold more sensitive to vincristine than wild-type mice, which is much less than the 128-fold reported previously (Johnson et al, 2001). It is unlikely that the different routes of administration in the two studies (i.p. vs i.v.) would explain this discrepancy. In our experience, the clearance of hydrophobic drugs from the peritoneal cavity of mice occurs rapidly. Moreover, both wild-type and Mdr1ab/Mrp1 TKO mice received vincristine by the same route, and the results for wild-type mice were consistent between the two studies. The difference may be explained by differences in drug elimination between animals in the two studies. In addition to elimination by excretion, metabolism is also a very important route for many drugs including vincristine. It has previously been shown that Mdr1a KO mice maintained in our institute have a higher expression of cytochrome P450 isoenzymes than mice of similar genotype kept in the United States (Schuetz et al, 2000). Consequently, clearance of vincristine might occur more rapidly in our populations of Mdr1ab DKO and Mdr1ab/Mrp1 TKO mice, rendering them less susceptible to vincristine.
To eliminate the effects of reductions in body clearance of vincristine on bone marrow toxicity, we have used a bone marrow transplantation model yielding chimeras that were of identical wild-type genotype, except for their bone marrow progenitor cells. The course of the WBC counts in the Mdr1ab/Mrp1 TKO chimeras closely resembled the pattern that is usually observed in patients experiencing chemotherapy-induced myelotoxicity. The nadir and return to baseline levels occurred somewhat more abruptly, but this is most likely due to the fact that most physiological processes in mice proceed at a higher velocity than in humans. Interestingly, the nadir was about similar in Mrp1 KO chimeras, but there was a very strong rebound in WBC counts by day 7. This difference between Mrp1 KO and Mdr1ab/Mrp1 TKO chimeras may be due to the fact that the relative expression of ABC transporters appears to vary along the lineage from uncommitted stem cells to mature blood cells. In mice, CD34− sorted SP cells appear to represent a more primitive subpopulation of progenitor cells than CD34+ cells (Osawa et al, 1996) and these cells contain relatively high levels of Bcrp1 and Mdr1 mRNA, whereas Mrp1 appears to be higher in the murine CD34+ subpopulation of cells (Zhou et al, 2001). The absence of Mrp1 will make this population of CD34+ progenitor cells more vulnerable to vincristine. A single bolus of a high dose of vincristine given to Mrp1 KO chimeras may significantly reduce the numbers of these precursor cells, thus eliminating part of the maturing blood cells from the pipeline. The decline in WBC will trigger signalling to induce a compensatory wave of haematopoiesis and because the population of primitive CD34− precursor cells is probably not so much affected due to protection by P-gp, this can still occur effectively. In Mdr1ab/Mrp1 TKO chimeras, however, this more primitive population of CD34− cells is no longer protected by P-gp and may therefore be more vulnerable to vincristine, thus causing further delay in WBC recovery.
In this study, we have shown that a single dose of vincristine resulted in a clear but nonlethal bone marrow toxicity, which was Mrp1 and P-gp dependent. Repeated dosing or continuous infusion of this G2–M cell cycle specific drug might have resulted in greater bone marrow toxicity. More pronounced cytotoxic effects on these primitive precursor cells have been shown in a recent study, where irradiated mice that were transplanted with mixtures of wild-type and Bcrp1 KO donor bone marrow were challenged with mitoxantrone (Zhou et al, 2002). After five daily repeated injections of 2 mg of mitoxantrone per kilogram, the relative contribution of Bcrp1 null cells in the peripheral blood myeloid and lymphoid compartment declined to very low values and remained low during the many weeks afterwards. In this case with repeated dosing of mitoxantrone, it appears that the majority of primitive Bcrp1 null stem cells have been eradicated and have been replaced by wild-type cells. Given that mitoxantrone is also a substrate of P-gp, we expect that the effect of this drug may be even greater in bone marrow cells with a compound deletion of the Mdr1ab and Bcrp1 alleles.
In conclusion, although the physiological function of P-gp, Mrp1 and other ABC-transporters in haematopoietic progenitor stem cells is still conjectural, it is clear that they protect the cells against chemotherapy-induced injury. Inhibition of their function, for example, as part of drug regimens aimed to sensitise drug-resistant tumour cells, may thus result in enhanced myelotoxicity. However, the fact that multiple transporters with (partly) overlapping substrate specificities are present appears to be a safety mechanism that may render this a relatively infrequent complication.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Allen JD, Brinkhuis RF, van Deemter L, Wijnholds J, Schinkel AH (2000) Extensive contribution of the multidrug transporters P-glycoprotein and Mrp1 to basal drug resistance. Cancer Res 60: 5761–5766
Borst P, Evers R, Kool M, Wijnholds J (1999) The multidrug resistance protein family. Biochim Biophys Acta 1461: 347–357
Boven E, Jansen WJM, Hulscher TM, Beijnen JH, van Tellingen O (1999) The influence of P170-glycoprotein modulators on the efficacy and the distribution of vincristine as well as on MDR1 expression in BRO/mdr1.1 human melanoma xenografts. Eur J Cancer 35: 840–849
Chaudhary PM, Roninson IB (1991) Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell 66: 85–94
Hipfner DR, Deeley RG, Cole SP (1999) Structural, mechanistic and clinical aspects of MRP1. Biochim Biophys Acta 1461: 359–376
Johnson DR, Finch RA, Lin ZP, Zeiss CJ, Sartorelli AC (2001) The pharmacological phenotype of combined multidrug-resistance mdr1a/1b- and mrp1-deficient mice. Cancer Res 61: 1469–1476
Jonker JW, Buitelaar M, Wagenaar E, van der Valk MA, Scheffer GL, Scheper RJ, Plösch T, Kuipers F, Oude Elferink RPJ, Rosing H, Beijnen JH, Schinkel AH (2002) The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci USA 99: 15649–15654
Jonker JW, Smit JW, Brinkhuis RF, Maliepaard M, Beijnen JH, Schellens JH, Schinkel AH (2000) Role of breast cancer resistance protein in the bioavailability and fetal penetration of topotecan. J Natl Cancer Inst 92(20): 1651–1656
Kim M, Turnquist H, Jackson J, Sgagias M, Yan Y, Gong M, Dean M, Sharp JG, Cowan K (2002) The multidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin Cancer Res 8: 22–28
Lin ZP, Johnson DR, Finch RA, Belinsky MG, Kruh GD, Sartorelli AC (2002) Comparative study of the importance of multidrug resistance-associated protein 1 and P-glycoprotein to drug sensitivity in immortalized mouse embryonic fibroblasts. Mol Cancer Ther 1: 1105–1114
Lorico A, Rappa G, Finch RA, Yang D, Flavell RA, Sartorelli AC (1997) Disruption of the murine MRP (multidrug resistance protein) gene leads to increased sensitivity to etoposide (VP-16) and increased levels of glutathione. Cancer Res 57: 5238–5242
Osawa M, Hanada K, Hamada H, Nakauchi H (1996) Long-term lymphohematopoietic reconstitution by a single CD34− low/negative hematopoietic stem cell. Science 273: 242–245
Rao VV, Dahlheimer JL, Bardgett ME, Snyder AZ, Finch RA, Sartorelli AC, Piwnica-Worms D (1999) Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood–cerebrospinal–fluid drug–permeability barrier. Proc Natl Acad Sci USA 96: 3900–3905
Scharenberg CW, Harkey MA, Torok-Storb B (2002) The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 99: 507–512
Schinkel AH, Mayer U, Wagenaar E, Mol CAAM, van Deemter L, Smit JJM, van der Valk MA, Voordouw AC, Spits H, van Tellingen O, Zijlmans JMJM, Fibbe WE, Borst P (1997) Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA 94: 4028–4033
Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CAAM, van der Valk MA, Robanus-Maandag EC, te Riele HP, Berns AJM, Borst P (1994) Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood–brain barrier and to increased sensitivity to drugs. Cell 77: 491–502
Schinkel AH, Wagenaar E, Mol CAAM, van Deemter L (1996) P-glycoprotein in the blood–brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest 97: 2517–2524
Schuetz EG, Umbenhauer DR, Yasuda K, Brimer C, Nguyen L, Relling MV, Schuetz JD, Schinkel AH (2000) Altered expression of hepatic cytochromes P-450 in mice deficient in one or more mdr1 genes. Mol Pharmacol 57: 188–197
Sparreboom A, van Asperen J, Mayer U, Schinkel AH, Smit JW, Meijer DKF, Borst P, Nooijen WJ, Beijnen JH, van Tellingen O (1997) Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci USA 94: 2031–2035
Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC (1987) Cellular localization of the multidrug resistance gene product in normal human tissues. Proc Natl Acad Sci USA 84: 7735–7738
van Asperen J, Schinkel AH, Beijnen JH, Nooijen WJ, Borst P, van Tellingen O (1996) Altered pharmacokinetics of vinblastine in Mdr1a P-glycoprotein-deficient mice. J Natl Cancer Inst 88: 994–999
Wijnholds J, de Lange ECM, Scheffer GL, van den Berg DJ, Mol CAAM, van der Valk M, Schinkel AH, Scheper RJ, Breimer DD, Borst P (2000) Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood–cerebrospinal fluid barrier. J Clin Invest 105: 279–285
Wijnholds J, Evers R, van Leusden MR, Mol CAAM, Zaman GJ, Mayer U, Beijnen JH, van der Valk M, Krimpenfort P, Borst P (1997) Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat Med 3: 1275–1279
Wijnholds J, Scheffer GL, van der Valk M, van der Valk P, Beijnen JH, Scheper RJ, Borst P (1998) Multidrug resistance protein 1 protects the oropharyngeal mucosal layer and the testicular tubules against drug- induced damage. J Exp Med 188: 797–808
Workman P, Twentyman P, Balkwill F, Balmain A, Chaplin D, Double J, Embleton J, Newell D, Raymond R, Stables J, Stephens T, Wallace J (1998) United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) guidelines for the welfare of animals in experimental neoplasia (second edition). Br J Cancer 77: 1–10
Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP (2002) Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci USA 99: 12339–12344
Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7: 1028–1034
Acknowledgements
We thank Marije Buitelaar for performing the PCR assays, Ton Schrauwers for expert biotechnical assistance and Drs AH Schinkel, PR Wielinga and P Borst for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
van Tellingen, O., Buckle, T., Jonker, J. et al. P-glycoprotein and Mrp1 collectively protect the bone marrow from vincristine-induced toxicity in vivo. Br J Cancer 89, 1776–1782 (2003). https://doi.org/10.1038/sj.bjc.6601363
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601363
