
Abstract
Hypomyelination and congenital cataract is a recently reported autosomal recessive white matter disorder characterized by hypomyelination of the central and peripheral nervous systems, progressive neurological impairment and congenital cataract and caused by mutations in gene DRCTNNB1A. Here we report a large intragenic deletion that does not lead to congenital cataract in all of the patients in an afflicted family. The clinical phenotypes described for five patients broaden the phenotype of the disease and indicate that congenital cataract is not an essential criterion for differential diagnosis.
Similar content being viewed by others

Introduction
Hypomyelination and congenital cataract (HCC) is a new white matter disorder characterized by hypomyelination of the central and peripheral nervous systems, progressive neurological impairment and congenital cataract. This disease was reported recently by Zara et al,1 who also identified the gene responsible as DRCTNNB1A. Here we report genetic analyses in a large consanguineous family from Eastern Turkey and detailed clinical features in one of the five affected individuals. The clinical evaluation, together with identification of a large deletion encompassing two exons of DRCTNNB1A, allowed us to assign the disorder as HCC. However, we report on one patient who developed bilateral cataract at 9 years of age and another patient with persistent unilateral cataract at 12 years of age.
Methods
Subjects
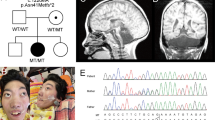
The large consanguineous family who applied to us for a genetic study had five affected members, three of whom survived 10, 13 and 16 years, respectively (Figure 1). This study has been approved by the Committee on Research with Human Participants at Boğaziçi University.

Partial pedigree diagram and haplotype analysis at 7p21.3–p12.3. Disease haplotype is boxed.
Molecular studies
A genome scan2 for the family at NHLBI Mammalian Genotyping Service (contract no. HV48141), followed by statistical analysis and fine-mapping identified a 9.02-Mb gene locus between markers D7S493 and D7S632 (Figure 1). This locus included gene DRCTNNB1A reported later as responsible for HCC by Zara et al.1 We analyzed 10 of the 11 coding exons of the gene in our patients. Exon 11 was not analyzed, since it is part of a low-expressed isoform. The sequences of intronic primers flanking each exon together with splice junctions were as reported,1 except for exons 8 and 12. The details of the analyses are provided online (Supplementary Methods Online).
Results
Clinical studies
A detailed clinical investigation was available for one patient only, whereas for the other four patients clinical information was partial and no instrumental investigations were available. The clinical manifestations of all five patients were similar to those described in Zara et al,1 except that patient 504 (Figure 1, the proband) never walked even with support; in patient 509 cataract did not develop until the age of 9 years; and in patient 505, cataract was congenitally present only unilaterally. We briefly describe here the findings for the proband. His medical records reported operations for bilateral cataract in the first month of life. He achieved normal development until 1 year of age, and from then on both motor and mental deterioration began. At the age of 5 years, he could not sit without support and could not walk even with support. Moderate mental retardation was observed. Diffuse and symmetrical deep white matter changes as shown in T2-weighted image hyperintensities and T1-weighted image hypointensities were seen in supratentorial structures (Supplementary Figure 1 online). Paraventricular white matter comprising the temporal horns, internal and external capsules, corona radiata and centrum semiovale were involved symmetrically in both the anterior and posterior parts of the cerebral hemispheres. Slight cerebral atrophy was also observed. Low nerve conduction velocities measured at age 2.4 years, ranging from 20 to 30 m/s in sensory and motor nerves, accompanied by prolonged F-response and by denervation potentials on EMG, verified a sensory–motor peripheral demyelinating neuropathy. Available clinical findings for the affected children are summarized in Table 1. Patients 502 and 505 were deceased at the time of the study; therefore, DNA and detailed clinical reports from these individuals were not available. Patient 502 died at the age of 2 years during the operation for the recurrent cataract. The parents of patient 505 stated that she had congenital cataract only unilaterally, and whether she later developed it in the other eye is not known. Her death at the age of 12 years was attributed to the disease.
Molecular studies
The disease gene was linked to a 9.02-Mb region with a maximum multipoint LOD score of 4.41 in the interval D7S2510–D7S2492 (Supplementary Figure 2 online). This region contained 92 genes, including DRCTNNB1A, which was responsible for HCC.1 We analyzed exons 1–7, 10 and 12 by SSCP, but did not observe any aberrant pattern in the three patients available for molecular study. However, exons 8 and 9 were refractory to PCR amplification, suggesting the presence of a homozygous deletion mutation. Amplification using intronic primers P1_delF and P1_delR yielded an approximately 2-kb fragment rather than the 6.3-kb fragment observed from the normal individuals, supporting the presence of a deletion (Supplementary Figure 3a online). Sequence analysis of the small fragment identified a deletion of 4084 nt encompassing exons 8 and 9 (Supplementary Figure 3b on line). The region of the deletion is flanked by an 82- and 83-bp direct repeat. In the mutant allele, a 4001-bp region together with the adjacent halves of the flanking repeat copies had been lost. This mutation c.531-439_743+348del is predicted to lead to the shift of the translational reading frame at the beginning of exon 10 and thus the truncation of the protein, 5 codons into the exon.
Discussion
Linkage to 7p together with close clinical resemblance to the disorder reported recently by Zara et al1 led us to assign the disorder in our patients as HCC. We subsequently showed that our patients carried in the disease gene a homozygous large deletion that apparently resulted from an unequal crossover at the direct copies of an imperfect repeat. The mutation would cause skipping of exons 8 and 9, and due to the 1-nt shift in the translational reading frame, is predicted to lead to a premature stop codon at position 214. The truncation deletes 308 amino-acid residues from the C terminus of the native 521 amino-acid protein. This mutation c.531-439_743+348del is predicted to result in absence of the protein since the deleted region spans more than half the coding region, including amino acids 230–248, which comprise one of the two putative transmembrane segments reported by Zara et al.1 The mutations reported in the mentioned study are either splice site (c.51+1G>A and c.414+1G>T) or missense (c.158T>C). It is difficult to speculate on the relative severities of the splicing and truncating mutations. All 10 patients in the previous report1 could walk with support until at least age 8 years, while none of our four patients who survived beyond age 2 years could walk with support past 6 years. However, while all patients in the former study had bilateral congenital cataracts, two of our patients did not: patient 509 developed cataracts at age 9 years and her sister (patient 505) had cataract seemingly only unilaterally until she died at the age of 12 years. Our findings add to the clinical variability reported already,1 in that congenital or bilateral cataract should not be considered a prerequisite for the diagnosis of HCC. Especially in isolated cases, bilateral or congenital cataract as a criterion for HCC would lead to misdiagnosis.
Accession codes
References
Zara F, Biancheri R, Bruno C et al.: Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat Genet 2006; 38: 1111–1113.
Weber JL, Broman KW : Genotyping for human whole-genome scans: past, present, and future. Adv Genet 2001; 42: 77–96.
Acknowledgements
We thank the family for participating in the study. This work was supported by the Turkish State Planning Organization. A Tolun was partially supported by the Turkish Academy of Sciences. SA Ugur was a fellow of the Scientific and Technological Research Council of Turkey. We gratefully acknowledge the genome scan for the family performed by NHLBI Mammalian Genotyping Service (Contract Number HV48141).
Author information
Authors and Affiliations
Corresponding author
Additional information
Web Resources
Accession numbers and URLs for data presented herein are as follows:
Marshfield Institute, http://research.marshfieldclinic.org/genetics/
(http://research.marshfieldclinic.org/genetics/Genotyping_Service/mgsver2.htm)
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for sequences from human [accession numbers NM_032581.2 and NP_115970.2])
NCBI Map Viewer, http://www.ncbi.nlm.nih.gov/mapview/
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Ugur, S., Tolun, A. A deletion in DRCTNNB1A associated with hypomyelination and juvenile onset cataract. Eur J Hum Genet 16, 261–264 (2008). https://doi.org/10.1038/sj.ejhg.5201935
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201935
Keywords
This article is cited by
-
Genes involved in leukodystrophies: A glance at glial functions
Current Neurology and Neuroscience Reports (2008)
