
Abstract
Epidemiological data, animal studies and interventional studies provide evidence for a potential chemopreventive effect of selenium during development of colorectal cancer. The human glycoprotein Selenoprotein P (SeP) contains up to 50% of plasma selenium content. SeP is expressed in the gastrointestinal tract and the liver, where its expression is downregulated by various proinflammatory cytokines (Il1β, TGFβ, IFNγ). Previously, we have demonstrated dramatically reduced SeP expression in human colon adenomas. Here, we have identified a complex (A)4-C-(A)4-GG-(A)8-GCT-(TC)5-(T)17 (bp −429 to bp − 477) repeat structure within the SeP promoter and we have analysed this regulatory DNA sequence with respect to polymorphisms, genomic instability and functional relevance to promoter activity. As opposed to the (TC)5 variant we identified a novel (TC)3 polymorphism within this repeat in the general population, which conferred significantly reduced basal promoter activity to reporter gene constructs in HepG2 cells. Allelic distribution of this (TC)n element was similar in colon carcinoma patients and healthy controls. Additionally, we observed genetic instability within the (T)17 repeat motif in colon cancers of the mutator phenotype. This instability of the (T)17 repeat had no effect on basal promoter activity in reporter gene assays. In conclusion, we characterised a complex repeat structure within the SeP promoter that may be of functional relevance to SeP gene expression. Further studies on the effect of different SeP promoter genotypes on SeP protein expression and disease susceptibility are needed.
Similar content being viewed by others

Introduction
Human Selenoprotein P (SeP) is a plasma glycoprotein containing up to 50% of plasma selenium concentration.1 The human SeP gene is located on chromosome 5q312 and is composed of five exons.3 The gene for Selenoprotein P is unique among other eukaryotic selenoproteins as it contains 10–17 (10 in human SeP) in frame UGA codons in different species. The function of SeP is not known. The initial proposal that SeP acts as a selenium storage and transport protein due to its high content of selenocysteine residues and high plasma levels4 has not yet been confirmed. Recent studies suggest a function of SeP in antioxidative cell defense systems as it protects against diquat toxicity5 and neutralises peroxinitrite.6,7 Recently, 1850 bp of the gene promoter region were characterised and inhibitory regulation of SeP promoter activity, mRNA and protein expression by different cytokines like interleukin 1β, tumour necrosis factor α, transforming growth factor β1 and interferon γ has been demonstrated.3,8,9 Apart from liver, SeP-transcripts and -protein have been observed in various tissues.10,11,12 Recently, we were able to show a reduction of SeP mRNA as well as SeP-protein expression in colorectal adenomas, the benign precursors of colorectal cancers, as compared to adjacent normal colonic mucosa.13 However, the underlying pathophysiological mechanism and functional consequences of this altered SeP expression remain unclear.
As chromosomal region 5q was postulated to contain a tumour suppressor region and SeP expression is markedly decreased in colorectal adenomas, we further analysed and characterised the human SeP promoter region with respect to a complex DNA motif containing repetitive nucleotide sequences. Here, we identified both a novel polymorphism with functional relevance to promoter activity and demonstrate genetic instability of this complex repeat structure in colorectal tumors of the mutator phenotype.
Materials and methods
Samples, DNA preparation
A total of 56 randomly selected tissue samples of colorectal carcinomas and corresponding normal mucosa were analysed together with blood samples of 196 patients with colon cancer and 219 healthy volunteers. DNA from blood samples and from microdissected sections of paraffin embedded tissues were extracted by commercially available kits (Qiagen, Hilden; Roche Diagnostics, Mannheim, Germany).
SeP promoter fragment analysis
A 132 bp fragment of the SeP promoter, containing a TC(5) repeat followed by a T(17) repeat, was amplified using the forward primer 5′-CTAACAAAGGTCACACTGTG-3′ labelled with ABI-FAM (Applied Biosystems, Weiterstadt, Germany) at the 5′ end and the reverse primer 5′-GCTGAGCCAGCGAATAA-3′. PCR was performed in a volume of 50 μl containing 100 ng genomic DNA, 10 mM Tris (pH 8.4), 50 mM KCl, 1.5 mM MgCl2, 0.25 mM dNTPs, 5 pmol of each primer, and 0.5 units of Taq polymerase (Amersham, Braunschweig, Germany). Conditions for amplification were 5 min at 94°C for denaturation, 35 cycles at 94°C for 45 s, annealing at 52°C for 45 s and extension at 72°C for 45 s, with a final 72°C extension step of 60 min. Successful PCR-amplification was controlled by agarose gel electrophoresis and ethidium bromide staining. Fluorescent PCR products of the promoter repeat region were analysed using ABI PRISMTM 310 Genetic Analyzer and the Genescan software package (Applied Biosystems, Weiterstadt, Germany). To confirm identity of the PCR products and to investigate sequence differences of PCR products with altered fragment analysis patterns, PCR products generated with unlabelled primers were subcloned using TOPO TA Cloning® (Invitrogen) and analysed by cycle sequencing using ABI Prism® BigDye™ Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Weiterstadt).
Mutagenesis
A −1156 to −14 bp3 construct of the SeP promoter was used for introducing mutations according to the identified promoter alterations. Two different internal deletion constructs, construct TC3 containing a (TC)3-repeat instead of the wildtype (TC)5-repeat, and construct T13 containing a (T)13-repeat instead of the wildtype (T)17-repeat were generated from the wildtype vector by polymerase chain reaction based site-directed mutagenesis (Quik Change® site-directed mutagenesis kit, Stratagene, La Jolla, CA, USA). Mutated sequences were confirmed by cycle sequencing.
To exclude introduction of additional artificial mutations after site directed mutagenesis a fully sequenced DNA insert containing the mutated SeP promoter region was excised by Blp1 digest and re-introduced into the wildtype SeP promoter construct, which was used for transfection assays. Plasmids for transfection were prepared and purified using RPM® Rapid Pure Kit (BIO 101) according to the supplier's protocol.
Cell culture and transfection
The human hepatocarcinoma cell line HepG2 (ATCC, HB 8065) was cultured as described.3 For transfection studies, in a first series of experiments, cells were seeded in 12 well plates and transfected at 70% confluence. Transfection and determination of luciferase and β-galactosidase activity were performed as previously described.3 Reporter gene assays were independently repeated 12–18 times. In a second series, pilot studies with transfection at 20–30% confluence were performed using 6 cm culture dishes and repeated four times.
Statistical Analysis
The Chi2 Test was used for statistical analysis of the frequency of the promoter polymorphism.
Data of the luciferase reporter gene assays were analysed using the Wilcoxon Test.
Results
The human SeP promoter contains a motif with simple sequence repeats (SSR)
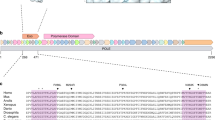
Inspection of the sequence of the human SeP promoter reveals a motif with repetitive DNA sequences with an (A)4-C-(A)4-GG-(A)8-GCT-(TC)5-(T)17 repeat (bp −429 to bp −477). Within this repeat structure a putative DNA binding site for the transcription factor nuclear factor of activated T cells (NFAT) has been identified (Figure 1).

The human Selenoprotein P promoter contains a complex sequence repeat motif. Diagram of the promoter (1810-bp 5′flanking exon 1) of the human Selenoprotein P gene with putative binding sites for SRY, NFAT AP1 and SP1 and details of the complex sequence repeat motif.
The human SeP promoter contains a polymorphic region within the repetitive DNA sequence motif
In blood samples of healthy volunteers and patients with colorectal cancer, analysis of the promoter motif of the human SeP promoter by fluorescence based fragment analysis indicated the occurrence of a polymorphism in this region. First, we found a homozygous allele pattern representing the known (TC)5(T)17 as confirmed by cycle sequencing of subcloned PCR products (Panel A, Figure 2). In addition, a second allele, four nucleotides smaller in size, was seen (Panel C, Figure 2). Sequence analysis revealed a novel polymorphism of the human SeP promoter with a (TC)3(T)17 repeat within this motif as compared to the wildtype sequence (Genbank ID Y12262). The sequence of the newly identified polymorphism was submitted to Genbank with the denoted accession number AF431733. Furthermore, samples with heterozygous peak patterns containing both the (TC)5(T)17 and the (TC)3(T)17 alleles of the SeP promoter were discovered by fragment analysis (Panel B, Figure 2) and subsequent cycle sequencing. These results indicate that the simple sequence repeat within the Sep promoter is polymorphic within the human genome.

Fragment analysis indicates a polymorphism within the TC-stretch of the complex repeat motif. Fragment analysis of the promoter sequence repeat motif shows pattern consistent with a long homozygous (TC)5/(TC)5 repeat (A), a long and short heterozygous (TC)5/(TC)3 repeat (B) and a short (TC)3/(TC)3 homozygous pattern (C).
The (T)17-single nucleotide repeat of the human SeP promoter is sensitive to genomic instability in tumors with mismatch repair deficiency
Fragment analysis of the SeP promoter motif in tissue samples of colorectal cancers and corresponding normal colon mucosa of the same patients displayed additional peaks in a fraction (10 of 56) of colon cancer tissues (Panel A, Figure 3) in contrast to corresponding normal colon mucosa, suggesting presence of genomic instability in this subgroup of tumors (Panel B, Figure 3). There was no significant difference in histological entity and tumour grading as well as age and sex of patients between tumour samples showing SeP promoter instability and those with stable SeP promoter sequences. Sequencing of subcloned PCR-products revealed that the instability is located within the (T)17-repeat (Figure 4), whereas no instability was observed within the (TC)5-repeat.

Genetic instability within the complex repeat motif of the SeP promoter. Representative fragment analysis of the SeP promoter motif in cancers of the mutator phenotype showing promoter instability as indicated by an altered pattern of the promoter repeat sequence in colorectal carcinoma (Panel B) compared to normal colon mucosa (Panel A) of the same patient is presented.

Genomic instability occurs within the (T)17 repeat of the SeP-promoter motif. Distribution of numbers n of (T)n repeat in a peripheral blood and colon carcinoma sample according to sequencing results from subcloned PCR products spanning the simple sequence repeat within the human SeP promoter. (PBL=peripheral blood leucocytes).
In order to exclude Taq polymerase amplification errors along the repeat motif, we amplified the repeat structure from DNA of cancer tissue and from blood of the same patient. The amplicons were subcloned and 46 subclones of the stable and 49 subclones of the unstable sample were analysed by sequence analysis. The results confirmed the occurrence of a broad spectrum of amplicons of different size in the unstable tumour samples ranging from (T)10 to (T)18, whereas amplicons of the corresponding blood DNA showed a T-repeat with a range of (T)15 to (T)18. The overall distribution of the T-repeat length within the subcloned and sequenced amplicons is shown in Figure 4.
These results confirm genomic instability within the Poly (T)-single nucleotide repeat of the human SeP promoter in a subset of colorectal cancers. This genetic instability within the (T)17-repeat of the SeP promoter was observed in 10 of 51 colon cancer samples with a (TC)5(T)17 motif. In contrast, no instability was seen in five colon cancers with a (TC)3(T)17 motif (Table 1). Colon cancers showing genetic instability within the promoter motif were identified as ‘MSI- high’ by standard methods, as defined by instability in at least 30 per cent of a consensus marker panel for MSI analysis (data not shown).14
Prevalence of the novel SeP promoter (TC)3 polymorphism is not elevated in colorectal cancer
We analysed the prevalence of this novel polymorphism in blood samples of 239 unrelated healthy volunteers in comparison to 196 patients with colon cancer. Among 239 healthy volunteers, the homozygous (TC)5/(TC)5 allele was found in 219 (91.6%) and the heterozygous (TC)5/(TC)3 allele in 20 (8.4%) resulting in a allele frequency of the (TC)5 allele of 0.96 and the (TC)3 allele of 0.04. None of the healthy volunteers but one of the colon cancer patients showed a homozygous (TC)3/(TC)3 allele. As compared to the healthy controls a similar allele distribution of the (TC)n-repeat was found in colon cancer patients indicating no relationship of this genotype to colon cancer development (Table 2).
Activity of the human SeP promoter is significantly reduced by the poly(TC)3-repeat polymorphism
Possible functional relevance of the (TC)3 polymorphism and the (T)17-repeat instability of the complex repeat element of the human SeP promoter was tested by luciferase reporter gene assays in of HepG2 cells, which were previously used for analysis of SeP promoter activity. We found no significant differences in basal promoter activity between the plasmids comprising the −1156 to −14 bp SeP promoter region and containing either the wildtype (TC)5(T)17 or the instability variant (TC)5(T)13 simple sequence repeat. In contrast, significant reduction of basal SeP promoter activity was observed for the promoter constructs containing the (TC)3(T)17-repeat as compared to the (TC)5(T)17-stretch when cells were transfected at 70% confluency (Figure 5). There was no significant change in promoter activity when cells were transfected at 20–30% confluency, possibly indicating proliferation-associated phenomena (data not shown). These results indicate that a polymorphism within the (TC)n-sequence repeat of the human SeP promoter significantly affects promoter transactivation.

A (TC)3-polymorphism within the SeP promoter affects basal promoter activity in HePG2 cells. Luciferase reporter gene assays using constructs comprising SeP-promoter wildtype ((TC)5(T)17), poly(TC)n-polymorphism ((TC)3(T)17) and poly(T)n-instability ((TC)5(T)13) were used for transfection of HepG2 cells. Luciferase activities of plasmids transfected in HepG2 cells are presented relative to the activity of the wildtype construct.
Discussion
Epidemiological data,15,16,17,18,19,20 animal studies21,22,23,24,25 and intervention studies in humans26 provide evidence for a potential chemopreventive effect of selenium in the development of colorectal cancer. Up to 50% of plasma selenium is contained in Selenoprotein P. SeP was shown to be expressed throughout the gastrointestinal tract including normal colonic mucosa.11 However, in colorectal adenomas SeP expression is markedly reduced,13 suggesting a possible role in prevention or promotion of colorectal carcinogenesis. Therefore, SeP may play an important role in colorectal tumorigenesis or its prevention.
Within the SeP promoter, a complex nucleotide sequence repeat structure containing an (A)4-GG-(A)8GCT-(TC)5-(T)17 motif is located between putative binding sites for different transcription factors. In this study, we investigated the relevance of alterations within this repeat structure of the SeP promoter for reduced SeP expression during colorectal carcinogenesis. We identified a novel polymorphism of the poly(TC) repeat of the promoter motif resulting in a (TC)3 repeat instead of a (TC)5 repeat. The observed polymorphism leads to a deletion of four bases within this short sequence repeat motif. Short sequence repeat motifs, also called simple sequence repeats (SSRs) are widely distributed throughout the genome.27 SSRs show a high rate of polymorphisms and are frequently used as genetic markers. In cells with defective mismatch repair, SSRs are prone to develop genetic instability, so called microsatellite instability, as a result of insufficient DNA mismatch repair of spontaneous mutations in repeat motifs due to strand mispairing during DNA replication.28 SSRs like poly(T) repeats29 and poly(TC) repeats30 can function as binding sites for regulatory proteins in upstream activation sequences. In several studies, deletion of SSRs from upstream promoter regions has been shown to reduce or eliminate transcriptional activity, whereas expansion of SSRs led to an increase of transcriptional activity, possibly due to alterations of binding of regulatory proteins.31 In this study, we also observed a significant reduction of basal promoter activity of plasmids containing the newly identified shorter (TC)3-polymorphism as compared as to the wildtype (TC)5-repeat in luciferase reporter gene assays in HepG2 cells. Differences in structure and spacing of the complex repeat elements or differences in the distance from the transcriptional start site as well as other factors may be responsible for reduction of promoter activity due to the (TC)3-polymorphism. Together, these results from promoter studies suggest that transcriptional inhibition of SeP expression due to the newly identified promoter polymorphism may lead to reduced protein levels of SeP.
Both the concentrations of Selenoprotein P and of total selenium in adult subjects vary considerably among different European regions,32 which cannot merely be explained by differences in nutritional selenium supply. In our study, reporter gene assays showed an association of the SeP promoter polymorphism (TC)3(T)17 with reduced basal promoter activity. Therefore, prevalences of different promoter genotypes may contribute to variations in Selenoprotein P plasma levels in different populations. This hypothesis remains to be proven.
To investigate the prevalence of the newly identified SeP promoter polymorphism in patients with colorectal cancers, we compared the frequency of the promoter polymorphism of 239 healthy volunteers to 196 colon cancer patients. No differences in the frequency of the promoter polymorphism could be demonstrated between the two groups. However, we observed the occurrence of alterations within the poly(T) repeat of the SeP promoter motif in a subgroup of colorectal cancers. Interestingly, no instability was observed within the (TC)n nucleotide repeats of the promoter motif. Further analysis of these tumours showed a high level of alterations in repetitive DNA sequences, also called microsatellite instability (MSI), which is characteristic for tumours of the mutator phenotype due to genetic alterations in genes of the DNA mismatch repair family. These results demonstrate, that the poly-(T)17-repeat of the SeP promoter is prone to genomic instability in tumors of the mutator phenotype.
In vitro, MSI can be increased by reactive oxygen species like hydrogen peroxide and diminished by antioxidative defense systems.33 It has been postulated that SeP may act as an antioxidative protein. Decreased expression of SeP in tumours through genetic alteration within the promoter may lead to an increase in DNA damage and thereby contribute to both tumour initiation and tumour progression.
The human SeP gene was mapped to chromosome 5q312, a chromosomal region which is linked to colorectal carcinogenesis (APC gene, 5q-deletion). In our study, 5 of 56 colon cancers showed a heterozygous allele of the TC-repeat and were therefore informative for analysis of loss of heterozygosity (LOH) of the SeP promoter. LOH as a possible cause of reduction of SeP was not observed. Because of the small number of informative samples, further studies using different markers seem to be necessary. In conclusion, we have newly identified an functionally important polymorphism within the SeP promoter and demonstrated genetic instability within the SeP promoter in colon cancers of the mutator phenotype. These observations may provide a molecular basis for variations in SeP expression due to genomic variability.
References
Harrison I, Littlejohn D, Fell GS . Distribution of selenium in human blood plasma and serum Analyst 1996 121: 189–194
Hill KE, Dasouki M, Phillips JA, Burk RF . Human selenoprotein P gene maps to 5q31 Genomics 1996 36: 550–551
Dreher I, Jakobs TC, Köhrle J . Cloning and characterization of the human selenoprotein P promoter. Response of selenoprotein P expression to cytokines in liver cells J Biol Chem 1997 272: 29364–29371
Motsenbocker MA, Tappel AL . A selenocysteine-containing selenium-transport protein in rat plasma Biochim Biophys Acta 1982 719: 147–153
Burk RF, Hill KE, Awad JA et al. Pathogenesis of diquat-induced liver necrosis in selenium-deficient rats: assessment of the roles of lipid peroxidation and selenoprotein P Hepatology 1995 21: 561–569
Arteel GE, Mostert V, Oubrahim H, Briviba K, Abel J, Sies H . Protection by selenoprotein P in human plasma against peroxynitrite- mediated oxidation and nitration Biol Chem 1998 379: 1201–1205
Sies H, Klotz LO, Sharov VS, Assmann A, Briviba K . Protection against peroxynitrite by selenoproteins Z Naturforsch 1998 53: 228–232
Mostert V, Dreher I, Köhrle J, Abel J . Transforming growth factor-beta1 inhibits expression of selenoprotein P in cultured human liver cells FEBS Lett 1999 460: 23–26
Hesse-Bähr K, Dreher I, Köhrle J . The influence of the cytokines Il-1beta and INFgamma on the expression of selenoproteins in the human hepatocarcinoma cell line HepG2 Biofactors 2000 11: 83–85
Hill KE, Burk RF . Selenoprotein P: recent studies in rats and in humans Biomed Environ Sci 1997 10: 198–208
Mörk H, Lex B, Scheurlen M, Dreher I, Schütze N, Köhrle J, Jakob F . Expression pattern of gastrointestinal selenoproteins–targets for selenium supplementation Nutr Cancer 1998 32: 64–70
Dreher I, Schmutzler C, Jakob F, Köhrle J . Expression of selenoproteins in various rat and human tissues and cell lines J Trace Elem Med Biol 1997 11: 83–91
Mörk H, Al-Taie OH, Bahr K et al. Inverse mRNA expression of the selenocysteine-containing proteins GI-GPx and SeP in colorectal adenomas compared with adjacent normal mucosa Nutr Cancer 2000 37: 108–116
Rodriguez-Bigas MA, Boland CR, Hamilton SR et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines [see comments] J Natl Cancer Inst 1997 89: 1758–1762
Schrauzer GN, White DA, Schneider CJ . Cancer mortality correlation studies–III: statistical associations with dietary selenium intakes Bioinorg Chem 1977 7: 23–31
Jansson B . Geographic mappings of colorectal cancer rates: a retrospect of studies, 1974–1984 Cancer Detect Prev 1985 8: 341–348
Dworkin BM, Rosenthal WS, Mittelman A, Weiss L, Applebee-Brady L, Arlin Z . Selenium status and the polyp-cancer sequence: a colonoscopically controlled study Am J Gastroenterol 1988 83: 748–751
Ghadirian P, Maisonneuve P, Perret C et al. A case-control study of toenail selenium and cancer of the breast, colon, and prostate Cancer Detect Prev 2000 24: 305–313
Russo MW, Murray SC, Wurzelmann JI, Woosley JT, Sandler RS . Plasma selenium levels and the risk of colorectal adenomas Nutr Cancer 1997 28: 125–129
Clark LC, Hixson LJ, Combs Jr GF, Reid ME, Turnbull BW, Sampliner RE . Plasma selenium concentration predicts the prevalence of colorectal adenomatous polyps Cancer Epidemiol Biomarkers Prev 1993 2: 41–46
Nelson RL, Abcarian H, Nelson TM et al. The effect of dietary selenium deficiency on acute colorectal mucosal nucleotoxicity induced by several carcinogens in the rodent Am J Surg 1996 172: 85–88
Jao SW, Shen KL, Lee W, Ho YS . Effect of selenium on 1,2-dimethylhydrazine-induced intestinal cancer in rats Dis Colon Rectum 1996 39: 628–631
El-Bayoumy K, Upadhyaya P, Chae YH et al. Chemoprevention of cancer by organoselenium compounds J Cell Biochem Suppl 1995 22: 92–100
Yan L, Yee JA, Li D, McGuire MH, Graef GL . Dietary supplementation of selenomethionine reduces metastasis of melanoma cells in mice Anticancer Res 1999 19: 1337–1342
Feng Y, Finley JW, Davis CD, Becker WK, Fretland AJ, Hein DW . Dietary selenium reduces the formation of aberrant crypts in rats administered 3,2′-dimethyl-4-aminobiphenyl Toxicol Appl Pharmacol 1999 157: 36–42
Clark LC, Combs GFJ, Turnbull BW et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial Nutritional Prevention of Cancer Study Group JAMA 1996 276: 1957–1963
Tautz D, Renz M . Simple sequences are ubiquitous repetitive components of eukaryotic genomes Nucleic Acids Res 1984 12: 4127–4138
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M . Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis Nature 1993 363: 558–561
Solomon MJ, Strauss F, Varshavsky A . A mammalian high mobility group protein recognizes any stretch of six A.T base pairs in duplex DNA Proc Natl Acad Sci USA 1986 83: 1276–1280
Yee HA, Wong AK, van de Sande JH, Rattner JB . Identification of novel single-stranded d(TC)n binding proteins in several mammalian species Nucleic Acids Res 1991 19: 949–953
Kashi Y, King D, Soller M . Simple sequence repeats as a source of quantitative genetic variation Trends Genet 1997 13: 74–78
Marchaluk E, Persson-Moschos M, Thorling EB, Akesson B . Variation in selenoprotein P concentration in serum from different European regions Eur J Clin Nutr 1995 49: 42–48
Jackson AL, Chen R, Loeb LA . Induction of microsatellite instability by oxidative DNA damage Proc Natl Acad Sci USA 1998 95: 12468–12473
Acknowledgements
We thank Tiemo Grimm, Institute of Human Genetics, Wuerzburg, for performing the statistical analysis of the promoter allele frequency. This work was supported by a grant of Deutsche Krebshilfe (I0-I492-Al I).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Al-Taie, O., Seufert, J., Mörk, H. et al. A complex DNA-repeat structure within the Selenoprotein P promoter contains a functionally relevant polymorphism and is genetically unstable under conditions of mismatch repair deficiency. Eur J Hum Genet 10, 499–504 (2002). https://doi.org/10.1038/sj.ejhg.5200811
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200811
Keywords
This article is cited by
-
Selenoproteins and oxidative stress-induced inflammatory tumorigenesis in the gut
Cellular and Molecular Life Sciences (2017)
-
Serum selenium and single-nucleotide polymorphisms in genes for selenoproteins: relationship to markers of oxidative stress in men from Auckland, New Zealand
Genes & Nutrition (2012)
-
Polymorphisms in the selenoprotein S and 15-kDa selenoprotein genes are associated with altered susceptibility to colorectal cancer
Genes & Nutrition (2010)
-
Selenoprotein P regulation by the glucocorticoid receptor
BioMetals (2009)
-
Polymorphism analysis of six selenoprotein genes: support for a selective sweep at the glutathione peroxidase 1 locus (3p21) in Asian populations
BMC Genetics (2006)
