
Abstract
Charcot-Marie-Tooth (CMT) disease is a typical example of a clinically and genetically heterogeneous disorder and, in most cases, is dominantly inherited and caused by a 1.5 megabase duplication on chromosome 17p11.2 containing the PMP22 gene. This is a non-lethal disease with a wide spectrum of severity, from asymptomatism to severe motor and sensory disability. Unpredictable degree of disability is usually the reason why prenatal diagnosis is required and must be addressed. Molecular procedures such as the use of polymorphic non microsatellite STRs, allowing very fast and reliable results even when requiring a gene dosage interpretation are now available and have been recently validated in post-natal diagnosis. Our results indicate that this approach is also the best-adapted method in case of prenatal diagnosis. Nevertheless, ethical considerations raised by prenatal diagnosis in CMT and more generally in non-lethal disorders remain to be actively considered. Here, we present our experience in genetic counselling, and address the psychological issues for 7 CMT at risk pregnancies. In five cases, a CMT1A duplication was evidenced; pregnancy was terminated in four of these cases and the parents from one affected foetus decided to pursue the pregnancy.
Similar content being viewed by others


Introduction
Charcot-Marie-Tooth disorders (CMT) represent a heterogeneous group of hereditary motor and sensory peripheral neuropathies (HMSN). Demyelinating and axonal forms have been described, and, to date, more than 30 loci have been identified.1 The most frequent form (CMT1A), corresponds to the stable inheritance of a 1.5 Mb duplication carried on chromosome 17p11.2–p12 containing the PMP22 gene and arising after unequal meiotic recombination.2,3,4,5
In the past decade, various technical procedures, ranging from Southern blot to FISH analysis, have been developed in order to detect the CMT1A duplication.6,7,8,9,10,11,12,13,14. In our experience, they may now be replaced by the use of non microsatellite polymorphic Short Tandem Repeats (STRs) for fluorescent PCR-based duplication diagnosis.15,16
Indeed, most of the procedures available for CMT1A duplication detection have been used in our laboratory, and our results clearly demonstrate that all duplications in CMT1A can be detected with a single fluorescent PCR approach and that qualitative results (presence of three alleles) are obtained for the entire set of duplications.
In the meantime, clinical variability in CMT1A, has to be considered. Although the most common phenotype is a chronic disease beginning in the second decade, early onset and severe disability are also observed.17,18,19 Since molecular diagnosis is available, prenatal diagnosis has been required by several at-risk families. In our center, the average of CMT1A duplications corresponds to 20 cases each year, and our series of prenatal diagnosis has been performed over a period of 2 years.
In this study, the experience of the pluridisciplinary prenatal diagnosis centre of Marseille for molecular evaluation and follow-up of five couples and seven cases of prenatal diagnosis of CMT1A is reported. We emphasize that a simple molecular testing has to serve as a support to address the essential questions being raised by prenatal diagnosis in heterogeneous disorders such as CMT1A; ethical and psychological aspects of prenatal diagnosis having to be actively discussed.
Materials and methods
Patients
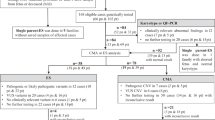
Our study includes five couples at-risk for CMT1A. Pedigrees are reported in Figure 1.

Pedigrees of five families requesting for prenatal diagnosis. Family 1 was partially reported previously.11 Haplotypes of STRs markers 4A, 9B, 9A are indicated. Families 2 and 3 have been explored separately, but they are represented on the same pedigree as the two affected husbands are brothers. In each family, haplotype is indicated in parenthesis when the number of copies (3) is deduced from the analysis of the relatives.
Consultations
A geneticist evaluated each couple at least twice in consultation, and familial history as well as the actual request was evaluated. Each case was discussed by the pluridisciplinary centre of prenatal diagnosis, including geneticists, paediatricians, and obstetricians, before any foetal sample was taken. One consultation or more with neurologists and at least one consultation with a psychiatrist were performed. Decision of carrying out the prenatal testing and anticipation of the modalities were then planned with the couple.
Samples
Genomic DNA from parents was extracted from peripheral blood lymphocytes under conditions accorded to standard procedures.20 DNA from foetuses was phenol extracted11 after trophoblastic biopsy taking place between 10 and 12 weeks of pregnancy. When available, amniotic cells were simultaneously sampled and cultured following standard protocols.21
Molecular analysis
A preliminary familial study was requested in all cases in order to define our molecular strategy and to check for the markers and family informativity. Three STRs markers (4 and 5 bp repeats) included within the CMT1A region were PCR amplified according to the recently reported protocol and primers15 modified as following: all markers were amplified individually and the forward primer was fluorescently labelled [6-carboxyfluorescein (FAM), 25 ng]. Products were resolved on an ABI310 automated sequencer (Applied Biosystems), and alleles were assigned with GENESCAN software (Applied Biosystems). Both allele numbers and allele's peak area were evaluated and results compared to the other technical procedures. Sequences from markers are issued from the following BACs identified with GenBank's access numbers: AC005703 (STR 4A), AC0013248 (STR 9A), AC0013248 (STR 9B) as reported.15
Results
Molecular testing
Among the seven prenatal tests that were performed, five foetuses (1F1, 1F2, 2F, 3F, 4F) were evaluated as carriers of the 17p11.2 duplication, while only two were unaffected (1F3 and 5F) (Figure 1); in family 1, duplication in parents and foetuses 1F1 and 1F2 was previously identified and results were reported in Bernard et al, 2000.11 Gene dosage analysis as well as CMT1A-REPs based Southern blot diagnosis8 and PCR assays were performed6,11 (data not shown). No discrepancy could be observed between the different technical procedures that were applied, including a standard microsatellite analysis.
STRs analysis was performed in all of the seven foetuses and related families and was informative in all cases (100%). Three alleles could be identified in all the affected foetuses for at least one marker out of the three included in the panel (Figure 2). For each affected foetus, markers retrieving 2 alleles sizes allowed to interpret the results on a gene dosage basis, and were then used as a result's confirmation. As expected from previous results, allele dosage was never observed among the non CMT1A-duplicated fetuses (Figure 2).

Non-microsatellite STR analysis after fluorescent PCR in families 1, 2 and 3. For each individual, specific alleles appear dark, while size markers appear clear. Family 1, STR 4A: Foetus 1F3 is heterozygous and no gene dosage can be observed. Familial analysis and gene dosage (1-1/5) observed in father (1.1) reveals that allele 1 (118 bp) is constitutive of the duplication (2 copies), while the normal father's chromosome is carrying one copy of allele 5 (128 bp). STR 9B and 9A: Both affected foetuses (1F1 and 1F2) harbour three distinct alleles. Foetus 1F3 is heterozygous for both markers with absence of allele dosage. STR 9B evidences the father's duplication (1.1), corresponding to the tandem of alleles number 6 (115 bp) and 7 (119 bp), while the normal chromosome carries only one copy of allele 6. The mother (1.2) has transmitted allele 5 (111 bp). Families 2 and 3: STR 9A identifies three distinct alleles in both affected fetuses 2F and 3F. STR4A and 9B are informative and gene dosage interpretation is conclusive for all individuals in these families. Results were correlated to those obtained with normal and CMT1A duplicated controls (data not shown). In each family, haplotype is indicated in parenthesis when the number of copies (3) is deduced from the analysis of the relatives.
Management of the families, issue of the pregnancies
For each couple, a reflection period was offered after the first request in order to ensure the decision before embarking in a prenatal diagnosis process. Couples 1 and 3 initially presented at the first genetics consultation before the pregnancy was engaged, while couples 2, 4, and 5 requested for a prenatal diagnosis within 4 weeks after beginning the pregnancy.
The fear of a severe impairment of the child to be born and the heaviness of the responsibility for having transmitted a disability for which biological diagnosis was available were the main arguments for requesting prenatal diagnosis. CMT1A duplication was found in five of the foetuses. In four cases, the parents decided to terminate the pregnancy, while in one case the couple eventually decided to carry on with the pregnancy (couple 3). In this latter case the couple had an interview with a psychiatrist after disclosure of the molecular results, while they had denied this offer before the analysis. Upon proposal, they also connected with the CMT-France patients association.
Discussion
A technical approach that is particularly suited in the case of prenatal diagnosis is reported. We applied this approach to several cases of prenatal diagnosis and our results show that using a set of three non-microsatellite STRs (4–5 bp repeats), to explore CMT1A duplications in prenatal diagnosis, satisfies all of the necessary criteria of liability, informativity, rapidity and ease of testing. In particular, no discrepancy was observed when compared to other methods. Finally, this procedure makes possible to detect any maternal contamination in the same experiment. Badano et al.16 reported a similar approach for CMT1A duplication diagnosis and thus diagnosed >99% of duplications by using multi-labelling and multiplex PCRs. Meanwhile, the easiness combined with an identical liability should make the 3-markers set more suitable for routine laboratory testing.
Beyond the necessity of an accurate technical approach, the main points of management for prenatal diagnosis of Charcot-Marie-Tooth disease are defined by the nature of the pathology itself. Charcot-Marie-Tooth disorders fall within the field of non-lethal dominant disorders, incompletely penetrant, with a high clinical variability. Thus, the probability of a mildly affected child being born has to be considered and confronted with the possibility of terminating the pregnancy.
Asymptomatism in CMT1A is observed in 10–15% of duplicated patients,18 while a severe disability with major neuromuscular impairment in childhood, bone deformities and other severe complications (respiratory failure, …) is only observed in very few cases.19,22
By itself the notion of handicap is questionable, as it is partly subjectively evaluated. The advice of neuro-pediatricians, as well as discussions with other parents of affected children might be useful for the requesting couple to progress toward their own decision. The intervention of psychiatrists or psychologists is sometimes difficult for the couple to accept, but may be crucial as the two parents, may not share the same feeling. Eventually, regardless of the final decision, psychological support and counselling are therefore of great importance to the couple during the entire process, as well as in the long-term follow-up of the families. The different steps of the process are summarized in Figure 3.

Schematic representation of the management of the couples requesting prenatal diagnosis in CMT1A.
Finally, the possibility for the couple to carry on with the pregnancy in case of positive CMT1A diagnosis must be addressed. Such option must be considered, since it happens once the couples have been provided with an invaluable information making possible an informed decision. In our experience, this situation happened in one case, essentially because the affected parent's history reflected important physical and/or psycho-social difficulties, making impossible for the couple to anticipate the possibility of assuming an affected child until both parents were, somehow, confronted with reality. In this case, prenatal diagnosis was requested by the couple as the only way to progress in their parent's role and was thus considered as a benefit. Such an experience could benefit to other couples experiencing a close situation related to non lethal and variable disorders.
Although it should not be considered as a failure in genetic counselling, this context is equivalent to pre-symptomatic diagnosis and deals, indeed, with the absence of informed consent for an affected individual when his parents have decided to carry on the pregnancy.
Pre-symptomatic diagnosis resulting from prenatal testing has been rarely reported, particularly concerning non-lethal, late onset and variable disorders. In case of neurodegenerative disorders, as in Huntington Disease, protocols have long time been designed to manage pre-symptomatic diagnosis and prenatal testing. Meanwhile, rare cases of ongoing pregnancies with unfavourable testing results have been reported, discussed and even criticized; however the right of the couple to make their own decision has never been questioned.23,24 The problem of prenatal genetic testing for genetic predisposition to cancer has also been raised and the results of several studies have markedly evidenced the heterogeneity of the points of view and managements, concerning the ethical, medical and psychological implications of such a testing.25,26 In CMT1A, the context slightly differs since neither the life duration nor the medical survey are concerned, and because the main question is the perception of the disability for the child to be born.
Nevertheless, debating on the stakes of prenatal diagnosis in these situations is difficult but unavoidable, and must be conducted under a pluridisciplinary light, particularly involving psychiatrists and psychologists. Supporting the couple and finding individual solutions should be the main objectives as we are convinced that there are no global ethical rules to apply systematically and the experience of prenatal diagnosis pluridisciplinary centres in non-lethal and variable disorders should then be systematically reported to ethical instances thus allowing advances in this complex domain.
Pre-implantation genetic diagnosis could solve many of the questions raised by prenatal diagnosis in case of variable disorders. This approach is challenging and most of the restrictions in using pre-implantation genetic diagnosis are due to technical limitations. One group, using microsatellite markers, succeeded in CMT1A.27 The use of non microsatellite highly polymorphic STRs allowing to retrieve three independent alleles as well as interpreting gene dosage when two different alleles are observed, indicates that pre-implantation genetic diagnosis for CMT1A should become technically easier in the near future. Testing the capability to accurately perform single cell PCRs from patients affected with CMT1A, by using the three STRs marker-set, is in process in our laboratory.
References
Meuleman J, Timmerman V, Nelis E, De Jonghe P . Molecular genetics of inherited peripheral neuropathies: who are the actors? Acta Neurol Belg 2000 100: 171–180
Lupski JR, Montes de Oca-Luna R, Slaugenhaupt S et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A Cell 1991 66: 219–232
Lupski JR . Charcot-Marie-Tooth polyneuropathy: Duplication, Gene dosage, and Genetic Heterogeneity Pediatric Research 1999 45: 159–165
Valentijn LJ, Bolhuis RA, Zorn I et al. The peripheral myelin genePMP22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A Nature Genet 1992 1: 166–170
Reiter LT, Murakami T, Koeuth T et al. A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element Nature Genet 1996 12: 288–297
Cudrey C, Chevillard C, Le Paslier D, Vignal A, Passage E, Fontes M . Assignment of microsatellite sequences to the region duplicated in CMT1A (17p12): a useful tool for diagnosis J Med Genet 1995 32: 231–233
Blair IP, Kennerson ML, Nicholson GA . Detection of Charcot-Marie-Tooth type 1A duplication by the polymerase chain reaction Clin Chem 1995 41: 1105–1108
Lopes J, LeGuern E, Gouider R et al. Recombination hot spot in a 3.2-kb region of the Charcot-Marie-Tooth type 1A repeat sequences: New tools for molecular diagnosis of hereditery neuropathy with liability to pressure palsies and of Charcot-Marie-Tooth type 1A Am J Hum Genet 1996 58: 1223–1230
Yamamoto M, Yasuda T, Hayasaka K et al. Locations of crossover breakpoints within the CMT1A-REP repeat in Japanese patients with CMT1A and HNPP Hum Genet 1997 99: 151–154
Stronach EA, Clarck C, Bell C et al. Novel PCR-based diagnostic tools for Charcot-Marie-Tooth type 1A and Hereditary neuropathy with liability to pressure palsies J Periph Nerv Syst 1999 4: 117–122
Bernard R, Labelle V, Negre P et al. Prenatal detection of a 17p11.2 duplication resulting from a rare recombination event and novel PCR-based strategy for molecular identification of Charcot-Marie-Tooth disease type 1A Eur J Hum Genet 2000 8: 229–235
Lebo RV, Martelli L, Su Y et al. Prenatal diagnosis of Charcot-Marie-Tooth disease type 1A by multicolor in situ hybridization Am J Med Genet 1993 47: 441–450
Navon R, Timmerman V, Lofgren A et al. Prenatal diagnosis of Charcot-Marie-Tooth disease type 1A (CMT1A) using molecular genetic techniques Prenat Diagn 1995 15: 633–640
Kashork CD, Lupski JR, Shaffer LG . Prenatal diagnosis of Charcot-Marie-Tooth disease type 1A by interphase fluorescence in situ hybridization Prenat Diagn 1999 19: 446–449
Latour P, Boutrand L, Lévy N et al. Polymorphic short tandem repeats for diagnosis of the Charcot-Marie-Tooth 1A duplication Clin Chem 2001 47: 829–837
Badano JL, Inoue K, Katsanis N, Lupski JR . New polymorphic short tandem repeats for PCR-based Charcot-Marie-Tooth disease type 1A duplication diagnosis Clin Chem 2001 47: 838–843
Ionasescu V . Charcot-Marie-Tooth neuropathies: from clinical descriptions to molecular genetics Muscle and Nerve 1995 18: 267–275
Birouk N, Gouider R, Le Guern E et al. Charcot-Marie-Tooth disease type 1A with 17p11.2 duplication. Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases Brain 1997 120: 813–823
Garcia CA . A clinical review of Charcot-Marie-Tooth Ann NY Acad Sci 1999 833: 69–76
Maniatis T . Molecular Cloning: A Laboratory Manual New York: Cold Spring Harbor Laboratory Press 1989
Bui TH, Iselius L, Lindsten J . European collaborative study on prenatal diagnosis: mosaicism, pseudomosaicism and single abnormal cells in amniotic fluid cell cultures Prenat Diagn 1984 1: 145–162
Dematteis M, Pepin JL, Jeanmart M, Deschaux C, Labarre-Vila A, Levy P . Charcot-Marie-Tooth disease and sleep apnoea syndrome: a family study Lancet 2001 357: 267–272
impson SA, Harper PS . Prenatal testing for Huntington's disease: experience within the UK 1994-1998 J Med Genet 2001 38: 333–335
Tolmie JL, Davidson HR, May HM, McIntosh K, Paterson JS, Smith B . The prenatal exclusion test for Huntington's disease: experience in the west of Scotland, 1986-1993 J Med Genet 1995 32: 97–101
Lodder LN, Frets PG, Trijsburg RW, Meijers-Heijboer EJ, Klijn JG, Niermeijer MF . Attitudes towards termination of pregnancy in subjects who underwent presymptomatic testing for the BRCA1/BRCA2 gene mutation in The Netherlands J Med Genet 2000 37: 883–884
Levy M, Richard S . Attitudes of von Hippel-Lindau disease patients towards presymptomatic genetic diagnosis in children and prenatal diagnosis J Med Genet 2000 37: 476–478
De Vos A, Sermon K, Van de Velde H et al. Pregnancy after preimplantation genetic diagnosis for Charcot-Marie- Tooth disease type 1A Mol Hum Reprod 1998 4: 978–984
Acknowledgements
We are grateful to the patients, families and the CMT-France association who made this work possible, Michel Sokolowski for psychiatric evaluation of the requesting couples, Claude Desnuelle, Jean-Claude Lambert, Jean Pouget, Brigitte Chabrol for helpful collaboration. Amandine Boyer was supported by a grant from the Lion's club Marseille doyen. This work was supported by the Assistance Publique–Hôpitaux de Marseille.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bernard, R., Boyer, A., Nègre, P. et al. Prenatal detection of the 17p11.2 duplication in Charcot-Marie-Tooth disease type 1A: necessity of a multidisciplinary approach for heterogeneous disorders. Eur J Hum Genet 10, 297–302 (2002). https://doi.org/10.1038/sj.ejhg.5200804
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200804
Keywords
This article is cited by
-
Provision and quality assurance of preimplantation genetic diagnosis in Europe
European Journal of Human Genetics (2008)
