
Abstract
The activation of caspases is the principal event in the execution of apoptosis. Initiator caspases are activated through an autocatalytic mechanism often involving dimerisation or oligomerisation. In Drosophila, the only initiator caspase DRONC, is tightly inhibited by DIAP1 and removal of DIAP1 permits activation of DRONC by the Drosophila Apaf-1-related killer, ARK. ARK is proposed to facilitate DRONC oligomerisation and autoprocessing at residue E352. This study examines whether autoprocessing of DRONC is required for its activation and for DRONC-mediated cell death. Using purified recombinant proteins, we show here that while DRONC autocleaves at residue E352, mutation of this site did not abolish enzyme activation, DRICE-induced cleavage of DRONC or DRONC-mediated activation of DRICE. We performed a detailed mutational analysis of DRONC cleavage sites and show that overexpression of DRONC cleavage mutants in Drosophila cells retain pro-apoptotic activity. Using an in vitro cell-free assay, we found ARK alone did not activate DRONC and demonstrate a requirement for an additional cytosolic factor in ARK-mediated DRONC activation. These results suggest that, similar to mammalian caspase-2 and caspase-9, the initial cleavage of DRONC is not essential for its activation and suggest a mechanism of ARK-mediated DRONC activation different from that proposed previously.
Similar content being viewed by others


Main
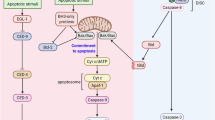
Apoptosis is mediated by a family of highly conserved cysteine proteases, known as caspases.1 Caspases exist as inactive zymogens and undergo intra-chain proteolytic cleavage to generate active large and small subunit heterodimers.1, 2 The crystal structures of some caspases demonstrate that the large and small subunits assemble to form an active heterotetramer.3, 4, 5 The prodomain of initiator caspases (caspase-2, -8, -9 and -10) is essential for docking proteins on oligomeric complexes to facilitate protein homodimerisation and subsequent autocatalytic processing to form active enzyme.6, 7, 8 The caspase prodomains contain protein–protein interaction motifs, such as the caspase recruitment domain (CARD) in caspase-2 and -9, and death effector domains in caspase-8 and -10.8 These domains allow dimerisation and/or interaction with adaptor proteins to facilitate initiator caspase activation.8 Once activated, the initiator caspases cleave various effector caspases, which form homodimers and target many vital cellular proteins for cleavage to execute the death of a cell.1, 9
In mammalian cells, apoptosis induced by stress (cytotoxic drugs, UV irradiation) results in permeabilisation of the outer mitochondrial membrane, causing release of cytochrome c into the cytosol.1, 10 This event mediates assembly of the apoptosome, composed of Apaf-1/dATP and cytochrome c. Once formed, the apoptosome recruits caspase-9 and mediates its autoproteolysis at residue D315.7, 11, 12, 13 The apoptosome is essential to mediate the dimerisation and stabilisation of active caspase-9.11, 12, 14, 15, 16 Furthermore, this dimeric caspase-9 has significantly enhanced activity compared to its monomeric form.17, 18 Interestingly, cleavage at D315 is not essential for caspase-9 activation by the apoptosome or for cleavage of caspase-3.16, 18, 19, 20 Active caspase-3 also cleaves caspase-9 at D330 in a feedback loop to remove an XIAP-binding IBM site, thereby alleviating XIAP inhibition of caspase-9 to allow sustained caspase activation.19
In Drosophila, the caspase-2/-9 homologue DRONC is required for specific developmental cell death pathways and stress-induced apoptosis21, 22, 23, 24, 25 and is activated by the Drosophila Apaf-1 homologue, ARK.26, 27, 28 ARK is structurally similar to Apaf-1 and in the presence of dATP, it forms an oligomer composed of eight ARK molecules, representing the fly apoptosome.29 While the ARK apoptosome does not contain or require cytochrome c, it can interact with the DRONC CARD.29 The recruitment of DRONC to the ARK apoptosome facilitates the initial DRONC cleavage at residue E352.30, 31 Cleavage at this glutamate residue is thought to enable formation of a stable DRONC homodimer and formation of an active catalytic site.30 Interestingly, during cell death the DRONC CARD is cleaved,32 which indicates that once activated DRONC does not require ARK to exert its downstream affects.
In light of caspase-9 activation studies, we explored whether proteolytic processing of DRONC was required for its enzymatic activity and for DRONC-mediated death in Drosophila cells. In this study, we have used recombinant purified DRONC proteins to elucidate the biochemical mechanism of DRONC activation. Our data indicate that while processing of DRONC is required for maximal activity, mutation of known target cleavage sites does not abolish DRONC activity, DRONC-mediated cleavage of DRICE or DRONC-induced cell death. We further show that ARK alone is not sufficient to activate DRONC in vitro. These results indicate that DRONC-activation shares some similarity to caspase-2 and -9 activation in mammals.
Results
DRONC processing is dependent on removal of DIAP1
DIAP1 is the main regulator of caspase activity in Drosophila cells and removal of diap1 by RNAi induces rapid caspase activation and cell death.33, 34 DIAP1 prevents apoptosis by directly inhibiting the caspases DRONC and DRICE, and cell death induced by diap1 depletion requires ARK and DRONC.34 To assess DRONC interactions in Drosophila cells, we transfected His6-tagged DRONC and pulled down protein with Ni-NTA agarose from untreated and cycloheximide-treated BG2 cells. In untreated control cells, we detected endogenous DIAP1 interacting with DRONC (Figure 1a). In apoptotic cells following cycloheximide treatment, we no longer detected DIAP1, but endogenous ARK now co-precipitated with DRONC, concurrent with DRONC processing to Pr2 at 36 kDa (Figure 1a).34 These results are consistent with previously reported data and demonstrate in vivo that removal of DIAP1 from DRONC during apoptosis allows DRONC interaction with ARK.

DIAP1 degradation is necessary for caspase activation in Drosophila cells. (a) Transfected DRONC-His6 protein from BG2 cells interacts with endogenous DIAP1 in untreated cells and with ARK in cycloheximide-treated cells. DRONC protein was pulled down with Ni-NTA agarose and interacting proteins immunoblotted for DIAP1 (top panel), ARK (middle panel) or DRONC (bottom panel). Lysates showing protein input are shown on the right. (b) BG2 cells treated with 10 μg/ml cycloheximide for 0–8 h in the absence or presence of MG132 (50 μM) and zVAD-fmk (50 μM). Lysates were immunoblotted with anti-DIAP1 antibody and caspase processing was detected by immunoblotting with anti-DRONC and anti-DRICE antibodies. Immunoblotting with anti-DRONC (p24) detects Pr1 in MG132-treated lysates. Immunoblotting with anti-cytochrome c was used as a protein loading control. (c) MG132 treatment of BG2 cells delays cycloheximide-induced apoptosis. Cell viability was determined by Trypan blue exclusion. zVAD-fmk (50 μM) was used to inhibit cycloheximide-induced cell death. Data were obtained from three separate experiments. Error bars indicate S.E.M. (n=3)
During apoptosis, DIAP1 is displaced from DRONC by RPR, HID and GRIM proteins, which act to decrease DIAP1 translation and induce DIAP1 degradation, thereby allowing caspase activation to proceed.35, 36 Treatment of Drosophila SL2 cells with apoptotic stimuli results in rapid removal of full-length DIAP1 protein35, 37 and this degradation is delayed by addition of the proteosome inhibitor MG132.37 We have used BG2 cells and confirmed that following treatment with cycloheximide, endogenous DIAP1 protein is degraded in 4 h, concurrent with cleavage of DRONC and DRICE processing at 6 h (Figure 1b). We used MG132 to assess the effect of sustained DIAP1 protein levels on caspase activation. MG132 treatment reduced DIAP1 degradation following cycloheximide treatment and importantly, resulted in delayed processing of both DRONC and DRICE and delayed cell death (Figure 1b and c). The addition of zVAD prevented caspase activation and cell death (Figure 1b and c) but did not prevent degradation of DIAP1. These results emphasise that during apoptosis, the proteosomal degradation of DIAP1 precedes caspase activation and is required for the processing and activation of both DRONC and DRICE.
Recombinant DRONC autoprocess at both glutamate and aspartate residues
The studies of mammalian caspase-2 and -9 have indicated that autoprocessing is not required for initiator caspase activation.14, 19, 38 This prompted us to investigate the association between DRONC cleavage and activation in Drosophila. Previous studies have shown that DRONC autocleavage at residue E352 to generate Pr1 is likely to be an initiating step to stabilise and activate the enzyme after which DRONC can further autocleave at E143.30, 31, 32, 34 We find that in cycloheximide-treated Drosophila BG2 cells, we only detect DRONC Pr1 following addition of MG132 and using a DRONC antibody raised against a polypeptide in the large subunit (p24) (Figure 1b).34 These results are consistent with previous reports and indicate that Pr1 may be degraded by the proteosome pathway.34 During stress-induced apoptosis in BG2 cells, DRONC is predominantly cleaved to a Pr2 (36 kDa) cleavage product (Figure 1b).26, 34 This can be mediated by DRICE cleavage at D135, which removes the DRONC CARD and is thought to represent fully activated DRONC protein.32
To further assess DRONC processing at residues E352, E143 or D135, we generated recombinant DRONC mutant proteins with an amino-terminal His6 tag (Figure 2a). We produced DRONC wild-type and processing-deficient mutant proteins in Escherichia coli and initially noted there was a temperature-dependent cleavage of full-length wild-type DRONC protein. While we detected complete processing of DRONC to p24 and p11 when protein was produced at 18°C (data not shown) and at 25°C, cleavage was reduced at 30°C and minimal at 37°C (Supplementary Figure 1). The p20 and p9 fragments seen are likely to be products generated from cleavage at alternative sites in the protein.

DRONC is processed at E352 and D135. (a) Schematic illustration of the DRONC mutants used in this study. All mutated residues are listed above wild-type DRONC. The aspartate and glutamate double mutants (D-dm, E-dm and DE/AA) comprise mutations in residues designated with crosses. (b) DRONC wild type (wt) and mutants were generated in E. coli and immunoblotted with either anti-His6 antibody to detect amino-terminal cleavage products or anti-DRONC to detect all carboxyl-terminal cleavage products. White line indicates where lanes have been joined. (c) A schematic representation of DRONC cleavage products
Mutation of the E143 site alone or together with E352A mutation (E-dm) was created to generate an uncleavable DRONC mutant. However, the E143A mutant did not affect DRONC processing and in fact mimicked the cleavage pattern of wild-type protein, indicating that DRONC is not cleaved at E143 (Supplementary Figure 1). Interestingly, while mutation of E352 to Ala prevented appearance of Pr1 at 40 kDa, this mutant was cleaved at alternative sites to generate p36 and p20 fragments when produced at 25°C or 18°C (Supplementary Figure 1 and data not shown). As expected, the catalytically inactive mutant C318G was not processed, indicating that the processing of DRONC in E. coli is dependent upon its catalytic activity.
To purify sufficient amounts of active DRONC, protein was produced in E. coli at 30°C. Purified recombinant DRONC activity was measured on fluorogenic peptide substrates over a 3 h time course to obtain maximal activity in our assays. DRONC exhibited activity on both VDVAD-AMC (7-amino-4-methylcoumarin) and DEVD-AMC substrates, and DRONC protein lacking the CARD (ΔCARD) displayed higher activity than full-length protein as previously reported (Supplementary Figure 2).33, 39 This activity was DRONC specific as a C318G mutant was inactive and pre-incubation of DRONC with DIAP1 effectively inhibited activity of wild-type DRONC but not ΔCARD protein (Supplementary Figure 2).
To investigate the autoprocessing of DRONC, we immunoblotted purified wild-type and mutant recombinant proteins with anti-His6 antibody to detect all amino-terminal cleavage products. We could detect Pr1 (40 kDa) product resulting from cleavage of DRONC at E352 and a p15/13 CARD fragment, which we assigned to cleavage at D135/D113 (Figure 2b). We also detected a p17 product, which was not a result of cleavage at E143. We tested if this p17 product was due to cleavage at nearby PSPE140, VGVVD166 or DGPE169 sites. However, mutation of any of these sites failed to prevent appearance of p17, indicating this product may be a consequence of nonspecific cleavage of recombinant protein (data not shown). To detect all carboxyl-terminal cleavage products, we used our DRONC (p14) antibody, which detected Pr1 and the p24 (D135–E352) large subunit (Figure 2b and c). Cleavage at E352 also released the small subunit, p11. The p20 fragment is likely a result of further cleavage of the large subunit (Figure 2b and c). As expected DRONC C318G was not processed. Figure 2c shows schematically, the various DRONC cleavage products observed in Figure 2b.
Processing of recombinant DRONC protein is not required for its activation
We analysed the catalytic activities of purified recombinant DRONC proteins using the fluorogenic caspase substrates VDVAD-AMC, DEVD-AMC and TQTE-AFC (7-amino-4-trifluoromethyl coumarin). We found that D135A and E140A mutants retained full VDVADase activity relative to wild-type DRONC protein (Figure 3a). The E143A, E352A and E-dm mutant proteins displayed 60–70% VDVADase and DEVDase activity of wild-type protein (Figure 3a and 3b). Importantly, while caspase activity on VDVAD and DEVD was slightly reduced, all mutants tested had significant catalytic activity compared to the inactive C318G mutant, indicating that mutation of DRONC cleavage sites does not abolish enzyme activity. Interestingly, the cleavage mutants tested had significantly reduced activity on TQTE-AFC compared to wild-type protein (Figure 3c). The D135A and E140A mutants possessed only 25% wild-type activity on TQTE substrate, indicating that cleavage of the CARD may be required for complete DRONC autoprocessing and maximal TQTE activity. We noted that the E143A mutant retained approximately 50% wild-type activity on TQTE substrate (Figure 3c), indicating that cleavage at E143 is not a requirement for DRONC autoprocessing. These results indicate that DRONC can be activated and cleave substrates' carboxyl-terminal aspartate residues without itself being proteolytic processed.

DRONC processing is not required for its catalytic activity. Catalytic activity of recombinant DRONC (100 nM) was assayed on (a) VDVAD-AMC, (b) DEVD-AMC and (c) TQTE-AFC substrates. Fluorescence was monitored over 3 h and the rate of substrate cleavage (pmol substrate cleaved per minute) was calculated and displayed graphically. Data were obtained from three separate experiments using three independent preparations of each protein. Error bars indicate S.E.M. *P<0.05, **P<0.01 and ***P<0.001 compared to wt DRONC protein activity
DRICE is processed by uncleaved full-length DRONC
To establish whether the processing-deficient DRONC mutants could process DRICE, we performed a cleavage assay using in vitro-translated (IVT) DRICE and DRONC proteins (Figure 4). As previously demonstrated, DRONC cleaves DRICE into p24 and p19 large and small subunits, respectively (Figure 4a).31 While the inactive C318G mutant was unable to mediate DRICE cleavage, the D113A and D135A mutants could still process DRICE (Figure 4a). Interestingly, a mutation of D324, which lies adjacent to the catalytic cysteine residue, was unable to cleave DRICE. We reasoned that mutation of D324 may alter the active site conformation and affect substrate binding.

Cleavage of DRICE and DRONC by various DRONC mutant proteins. In vitro-translated DRICE and DRONC proteins were incubated with the indicated recombinant DRONC mutants (1 μM). (a) Cleavage of DRICE by DRONC aspartate mutants (D113, D135 and D-dm). CARD represents DRONC (1-D135) fragment and is used as a control. (b) Cleavage of DRICE (top panel) and DRONC (middle panel) by DRONC glutamate mutants. Recombinant DRONC protein inputs are shown by immunoblotting with anti-DRONC antibody (lower panel)
We then assessed the ability of the glutamate cleavage mutants, E140A, E143A, E352A and E-dm, to process IVT DRICE and DRONC proteins. Consistent with our caspase activity results, we found that while each mutant displayed slightly less efficiency compared to wild-type protein, they were still able to cleave DRICE to p24 and p19 fragments (Figure 4b). Cleavage of IVT DRONC by wild-type recombinant DRONC protein resulted in Pr1 (40 kDa) and 38-kDa protein fragments. This cleavage was still detected when using the E143A mutant and partial cleavage was detected with D135A and E140A mutants (Figure 4b). Consistent with our TQTE caspase activity data, the E352A mutant was unable to cleave IVT-DRONC (Figure 4b). These data infer that DRONC can process and activate caspases without autoprocessing at E352, E143 or D135 sites.
DRONC processing at E352 is not essential for cell death
To investigate the importance of DRONC autocleavage in Drosophila cells, we transfected our DRONC mutant constructs (Figure 2a) into BG2 or SL2 cells and assessed their cleavage by immunoblotting (Figure 5a). Each of the mutant DRONC proteins expressed in the Drosophila cells comprised both amino-terminal HA tag and carboxyl-terminal His6 tag to detect all amino- and carboxyl-terminal cleavage fragments following cycloheximide-mediated apoptosis. Our data confirmed that the processing of DRONC to Pr2 is a result of cleavage at residue D135 (Figure 5a).39, 40 The D135A mutant was still processed at D113, albeit with reduced efficiency. Interestingly, while mutation of both D113 and D135 (D-dm) also results in loss of full-length protein following cycloheximide treatment, no cleavage products are evident (Figure 5a). As cycloheximide inhibits protein synthesis, it appears that the D-dm-uncleavable DRONC is rapidly degraded following treatment, and indicates that cleavage at D135 may stabilise the active protein. As expected, mutation of the active site C318G prevented DRONC processing following cycloheximide treatment. Interestingly, mutation of nearby D324 residue results in accumulation of full-length protein during apoptosis; however, cleavage to Pr2 is still detected. In addition, while we detect accumulation of full-length mutant E352A protein following cycloheximide treatment, we still detect cleavage to Pr2, indicating that cleavage at this site is not essential for cleavage of DRONC at D135. We also used a DRONC (p24) antibody to assess whether we could detect additional internal cleavage products. We were able to detect a p24 band equivalent to D135–E352 fragment in both untreated and cycloheximide-treated lanes, but no additional cleavage products were detectable. The presence of p24 fragments in our untreated samples may be a result of DRONC activation due to overexpression. To detect amino-terminal cleavage fragments, we immunoblotted cell lysates with anti-HA antibody, and found we could only detect full-length DRONC protein in our assay, but not CARD at 15 kDa or Pr1 at 40 kDa.

Processing-deficient DRONC mutants induce apoptosis in Drosophila cells. Wild-type and mutant DRONC expression constructs were transfected into BG2 cells. (a) Transfected DRONC protein was pulled down with Ni-NTA agarose and protein immunoblotted with anti-DRONC (p14) and (p24) antibodies. Lysates were immunoblotted with anti-HA antibody. White lines indicate where gel lanes have been joined. (b) Cell death induced by overexpression of wild-type DRONC and its mutants. Transfected DRONC protein expression was assessed by immunoblotting with anti-DRONC antibody, and anti-cytochrome c antibody was used as a loading control. Data were obtained from three separate experiments. Error bars represent S.E.M. (n=3). Transfected protein levels are shown by immunoblot using anti-HA antibody and anti-cytochrome c immunoblot was used as a protein loading control
Processing-deficient DRONC mutants do not act as dominant negative inhibitors
Our results suggested that the apparent DRONC cleavage site mutants are still processed at D135 and may retain the ability to induce death in Drosophila cells. Only the catalytically inactive mutant should act as a dominant negative, as we have previously shown.39 To test this hypothesis in vivo, we expressed full-length wild-type and mutant DRONC proteins in BG2 cells and assessed cell death induced by DRONC overexpression by scoring cells for apoptotic morphology using a β-galactosidase reporter assay. As demonstrated in Figure 5b, wild-type DRONC protein induces 45% apoptosis following 24 h transfection. The expression of D113A, D135A, D-dm and E143A mutants induced cell death comparable to wild-type protein. The E352A and E-dm proteins were less effective than the wild-type protein (∼30% compared to 48%) but could still promote significant levels of apoptosis. As expected, the catalytically inactive C318G DRONC failed to induce apoptosis and mutation at D324 was also unable to induce apoptosis when overexpressed, even though this mutant was still processed at D135. These results suggest that either DRONC cleavage at D324 is essential for its activation or like C318G, D324A has reduced catalytic activity and suppresses DRONC activation. We assessed the expression levels of transfected protein by immunoblotting with anti-HA antibody and found all mutant proteins were expressed at comparable levels to wild-type DRONC (Figure 5b).
Full-length DRONC is active in Drosophila cells
To test whether full-length active DRONC can be detected in cells undergoing apoptosis, we used affinity labelling with biotin-conjugated DEVD or VAD peptides to capture active caspases.7 Protein lysates from untreated or cycloheximide-treated (for 4 h) BG2 cells were incubated with either biotin-DEVD-CHO or biotin-VAD-fmk, followed by pull down with streptavidin sepharose. Biotin-VAD captured a 50-kDa band corresponding to full-length DRONC in cycloheximide-treated lysates, but not in untreated lysates (Figure 6). We were unable to detect processed Pr1 and Pr2 bands; however, a faint p24 cleavage fragment, representing DRONC large subunit could be detected (Figure 6; lane 16). We reasoned that the inability to efficiently capture a cleaved DRONC fragment is due to transient activation of DRONC, which is difficult to capture from apoptotic lysates. In contrast, processed DRICE was efficiently captured by both biotin-DEVD and -VAD. Pre-incubation of extracts with zVAD could outcompete and effectively block the capture of caspases with biotin-VAD (Figure 6). These results indicate that full-length, unprocessed DRONC and p24 DRONC are active. Furthermore, these results demonstrate that processed DRICE is a dominant activated caspase in apoptotic Drosophila cells.

Capture of active DRONC in Drosophila cells. Cytosolic extracts from untreated or cycloheximide-treated BG2 cells were incubated at 27°C with or without biotin-DEVD-CHO (lanes 5, 6 and 13, 14) or biotin-VAD (lanes 7–10 and 15–18). Biotinylated proteins were captured with streptavidin sepharose. Unbound and captured proteins were separated on SDS-PAGE and immunoblotted with anti-DRONC (top panel) or anti-DRICE (bottom panel) antibodies. zVAD (50 μM) was pre-incubated with cytosolic extracts prior to the addition of bVAD as a binding competitor (lanes 9, 10 and 17, 18). Protein lysate input is shown in lanes 1–2
ARK is not sufficient to activate DRONC
To reconstitute DRONC activation in vitro, we expressed and purified recombinant DRONC and ARK proteins in SF21 cells and assessed whether ARK could activate purified DRONC. We initially assessed ARK/DRONC apoptosome formation by incubating recombinant DRONC and ARK together in the presence of dATP and found that both proteins eluted together from a size exclusion gel filtration column in high molecular weight fractions 1–3 (>670 kDa) (Figure 7a). We next tested each fraction for DEVDase activity and found that the activity was not confined to fractions 1–3 containing ARK/DRONC and was seen across fractions 1–9, indicative of some background activity (Figure 7b). These results indicated that ARK/DRONC complex may be only partially active. To eliminate the possibility that IAP proteins may have co-purified with DRONC from SF21 cells, and were inhibiting DRONC activity, we added a HID peptide to our assay but did not detect any increase in DRONC activity (data not shown). Furthermore, the addition of either mammalian or Drosophila cytochrome c did not enhance DRONC/ARK complex formation or caspase activity (data not shown).

The apoptosome complex comprising ARK/dATP and DRONC is only partially active. (a) Recombinant ARK (150 nM) and DRONC (150 nM) proteins purified from SF21 cells were incubated alone or together in the presence of 5 mM dATP at 30°C for 30 min and then subjected to size exclusion chromatography. Protein from each fraction was analysed by immunoblotting with anti-DRONC and anti-ARK antibodies. (b) Individual fractions from DRONC/ARK/dATP reaction were assessed for caspase activity on DEVD-AMC. Data represent mean values obtained from the same fractions assayed in triplicate
We reasoned that perhaps an additional protein factor(s) was required for optimal protein folding and full caspase activation. In view of the structure of the ARK apoptosome, this protein factor is likely to be small to fit into its β-propeller regulatory region.29 To investigate whether other cytosolic components could induce ARK-mediated DRONC activation, we subjected BG2 protein lysates to size exclusion chromatography and pooled eluted fractions 29–38, containing proteins <20 kDa. We immunodepleted this protein pool of DRICE and cytochrome c to ensure that these proteins were not contributing to any caspase activity and added this depleted lysate to our in vitro reaction comprising purified ARK/dATP/DRONC. Interestingly, we found that this protein fraction could significantly enhance ARK-mediated DRONC DEVD-like activity (Figure 8a). Furthermore, this DRONC activity could be inhibited by addition of zVAD, confirming that the activity was caspase dependent (Figure 8a). When individual protein fractions 29–38 were incubated with ARK/dATP/DRONC, we found that fractions 29 and 30 (∼15–17 kDa) and fractions 36–38 (∼1–5 kDa) could enhance DRONC DEVD-like activity (Figure 8b).

Apoptosome-mediated DRONC activity requires an additional cytosolic factor. (a) Recombinant DRONC and ARK were incubated alone or together in the presence of dATP and a BG2 small molecular mass protein lysate, pooled from Superdex 200 fractions 29–38. DRONC activity was assessed on DEVD-AMC over 3 h and the rate of substrate cleavage was determined. Where indicated, zVAD (50 μM) was added to inhibit caspase activity. Error bars indicate S.E.M. (n=3). ***P<0.001 compared to activity of DRONC and ARK individually. (b) Individual protein fractions (nos. 29–38) isolated from Superdex 200 size exclusion chromatography were added to the in vitro reaction in (a) and caspase activity assayed on DEVD-AMC. The background activity of each fraction was deducted to give final RFU readings. In both experiments, error bars represent S.E.M. (n=3). (c) Cleavage of recombinant DRONC and IVT-DRICE protein by ARK/dATP is enhanced by addition of BG2 small molecular mass protein fraction. DRONC Pr1 and Pr2 fragments were detected by immunoblotting with anti-DRONC antibody. IVT-DRICE cleavage was detected by Phosphorimaging
To further assess whether this small molecular mass protein fraction could induce cleavage of DRONC and DRICE, we incubated recombinant DRONC or IVT-DRICE protein with ARK/dATP/BG2 lysate (Figure 8c). The BG2 protein fraction could not induce processing of DRONC or DRICE alone but together with ARK/dATP, we could detect DRONC Pr1 and Pr2 cleavage products (Figure 8c, top panel). In addition, while we detected partial cleavage of DRICE by DRONC/ARK/dATP, the addition of our BG2 protein fraction enhanced cleavage to p24 and p19 fragments (Figure 8c, lower panel). These results are consistent with our substrate cleavage data (Figure 8a), indicating that ARK alone may not be sufficient to activate DRONC and additional proteins/factors can enhance ARK-mediated DRONC and DRICE activation in Drosophila cells.
Discussion
This study investigated the requirement for DRONC autoprocessing in DRONC activation and cell death. Consistent with previous findings, we have demonstrated that DRONC autoprocessing occurs at E352 between the large and small subunits and also at D135 following the CARD but did not detect DRONC autocleavage at E140 or E143 sites. Importantly, our data indicate that DRONC cleavage is not necessary for enzyme activation and function. Furthermore, through its binding to bVAD we could detect full-length active DRONC in apoptotic cell extracts, suggesting that the initial activation of DRONC does not require its processing in vivo. This is similar to observations with mammalian caspase-238 and caspase-9,7, 14, 16, 18, 19, 20 which also do not require cleavage for initial activation of caspase zymogen.1, 8
Caspase activation in Drosophila cells is regulated by DIAP1 and the degradation of DIAP1 via the proteosome pathway is essential for activation of DRONC and DRICE. Structural studies have suggested that the activation of DRONC involves autocleavage at E352, which drives DRONC dimerisation and stabilisation of the active site.30 Interestingly, while mutation of E352 eliminates TQTE activity and DRONC autoprocessing, we have found that it does not abolish DRONC activity and cleavage of the effector caspase DRICE. Our findings differ from previously published data indicating that E352 mutations abolish its ability to mediate cell death in S2 cells.32 The discrepancy in results may partly be due to higher expression levels of our DRONC constructs, or perhaps higher levels of endogenous DRONC protein in BG2 cells. Nevertheless, our biochemical data are consistent with the cell death data.
It is clear from our data that, while DRONC autoproteolysis is not required for its initial activation, processing at E352 can further enhance DRONC activity by approximately 1.5-fold. These findings indicate that DRONC cleavage functions to provide maximal catalytic activation. The physiological relevance of DRONC TQTE-like activity is likely to primarily drive DRONC autocleavage and processing of DIAP1 at VQPE205 or other uncharacterised TQTE-like-containing substrates.40 However, it is clear from our data that this TQTE activity is not required for DRONC activation, DRONC-mediated processing of DRICE or for DRONC apoptotic function in vivo. The DRONC activity we detected on VDVAD and DEVD substrates is indicative of its ability to cleave effector caspases DRICE and DCP-1 at TETD sites.31 In addition, we detected recombinant DRONC p13/p15 cleavage fragments, which are equivalent to autocleavage at DESD113/DIVD135 sites. Taken together, our data indicate that DRONC can autoprocess carboxyl terminus of both glutamate and aspartate residues.31
During apoptosis DRICE can process DRONC at D135. We have demonstrated that mutation of D135 does not abolish DRONC activity and does not inhibit DRONC-mediated cleavage of DRICE. DRONC activation is thus unlikely to require DRICE-mediated proteolytic processing. Interestingly, as DRONC interaction with ARK is mediated by the CARD, cleavage of DRONC at D135 would release DRONC from the apoptosome.8 If we inhibit release of the CARD by mutating D135, this reduces TQTE-like activity and DRONC autoprocessing. Given that recombinant ΔCARD-truncated DRONC possessed enhanced activity compared to full-length DRONC, cleavage at D135 event may serve as a mechanism to stabilise the enzyme and provide maximal catalytic activity to perform its subsequent proteolytic functions. As DIAP1 inhibits DRONC by interacting with CARD, the removal of the CARD by cleavage at D135 may also serve an additional purpose in that it further prevents inhibition of active DRONC by DIAP1. This mechanism is analogous to the removal of the caspase-9 IAP-binding motif via caspase-3-mediated cleavage, to prevent further XIAP binding and inhibition of caspase-9 activity.19
Our observations are similar to that of mammalian initiator caspase-9 activation. While caspase-9 can autoprocess at D315 or be processed by caspase-3 at D330, this event is not necessary for initial caspase-9 activation.14, 16, 18, 19, 20 Instead, several studies have shown that binding of caspase-9 to the Apaf-1 apoptosome drives caspase-9 homodimerisation, which is sufficient to stabilise and activate caspase-9.11, 14, 17 Dimeric caspase-9 certainly has higher activity than monomeric caspase-9.41 In light of our data and from previously reported data, it is likely that in vivo, ARK acts as an adaptor to facilitate dimerisation of DRONC and stabilisation of an active conformation to enable downstream caspase activation. In vitro, high concentrations of recombinant DRONC protein may permit self-association and activation. Although cleavage is not required to activate DRONC, it is possible that autocleavage acts to stabilise active dimer formation such that once DRONC dissociates from the apoptosome, following cleavage at D135, it remains a dimer and retains its catalytic activity.32 Importantly, our findings show that unprocessed DRONC is active. As processed DRONC was not captured by bVAD in our system, it is feasible to suggest that fully processed DRONC may not be active and may be inactivated by dissociation from the apoptosome, similar to caspase-9.7 However, while we have shown that unprocessed DRONC is active in Drosophila cells, we cannot exclude that activation is a transient event and processed DRONC is not efficiently detected by biotin-VAD in our assay. It is expected that, like caspase-9, the processing of DRONC is required for optimal function and cleavage of downstream substrates.7
We explored apoptosome formation by gel filtration of recombinant ARK and DRONC proteins and demonstrated DRONC recruitment to ARK apoptosome in vitro. Unexpectedly, this complex had low activity and we did not observe DRONC processing when incubated with ARK alone. The finding that a protein factor of small molecular mass enhances ARK-mediated DRONC activity indicates that like mammalian Apaf-1, ARK may require an additional cytosolic factor, other than cytochrome c, to form an active conformation for DRONC binding. Importantly, this data show that ARK is not sufficient to activate DRONC alone. These findings, importantly indicate that ARK activation does in fact share similarity to the mammalian apoptosome. Further analysis is required to characterise this activating factor and also to assess whether, like activation of caspase-9, the ARK apoptosome acts to stabilise DRONC so that it can more readily bind effector caspases (DRICE, DCP-1).7, 17
In summary, this study demonstrates that processing of DRONC is not necessary for its initial activation. While uncleavable forms of DRONC had reduced TQTEase activity, they were still able to process the effector caspase DRICE and were able to mediate cell death when overexpressed in Drosophila cells. We have demonstrated that DRONC forms part of the ARK apoptosome in vitro but an additional, uncharacterised cytosolic factor is required to facilitate DRONC and DRICE activation by ARK. We are currently attempting to purify this factor. In view of the data presented here, we conclude that unprocessed DRONC is active and ARK-mediated DRONC autocleavage likely functions to stabilise its active site and further augment enzyme activity to improve efficiency of initiation of the caspase cascade.
Materials and Methods
Information on constructs, cell culture, recombinant protein expression and purification can be found in Supplementary material at www.nature.com/cdd.
Transfections
BG2 cells were transiently transfected with 1.5 μg HA-DRONC-His6 wild-type and mutant constructs together with 0.5 μg pIE1.4-LacZ reporter construct using Cellfectin reagent (Invitrogen). At 24 h following transfection, cells were fixed and stained in X-gal solution as previously described.39 Percent apoptosis was calculated by scoring >400 β-galactosidase-positive cells for apoptotic morphology.39 Where indicated, transfected HA-DRONC-His6 proteins were pulled down using Ni2+-NTA agarose (Qiagen).
Caspase assays
Fluorogenic substrate cleavage assays were carried out in caspase assay buffer comprising 0.1 M Hepes pH 7.5, 0.1% CHAPS, 10% sucrose, 10 mM dithiothreitol, 50 mM NaCl, 0.5 mM EDTA, supplemented with protease inhibitor mix (Roche). Assays contained 100 μM of either DEVD-AMC (Enzyme System Products) VDVAD-AMC (California Peptide Research) or 20 μM TQTE-AFC (CalBiochem) and either 20 μg of bacterial lysates or 150 nM of purified recombinant DRONC (wild-type or mutant protein), in a total volume of 100 μl. For in vitro cell-free assays, 150 nM DRONC and 150 nM DARK were incubated in the presence of buffer A containing 5 mM dATP, at 30°C for 2 h. Assays contained 1 μM DIAP1, 50 μM zVAD or 20 μg protein lysates where indicated. The cleavage of substrates was analysed on a Luminescence Spectrometer (Perkin-Elmer) (AMC excitation 360 nm, emission 460 nm; AFC excitation 400 nm, emission 520 nm).
35S-Protein cleavage assays
DRONC and DRICE cDNA were cloned into pCDNA3 expression vector (Invitrogen). Proteins were produced using a TNT-rabbit reticulocyte translation system (Promega) and incorporating 35S-methionine (Amersham Biosciences). 35S-labelled protein (4 μl) was incubated with 1 μM caspase in a volume of 40 μl in caspase assay buffer, for 3 h at 30°C. Proteins were resolved by 15% SDS-PAGE and transferred to PVDF membranes which were then exposed to a Phophor screen. 35S-Signals were visualised using the Typhoon 9410 and ImageQuant software (Amersham).
Immunoblotting
Protein lysates were prepared by resuspending cell pellets in PBS and boiled in an equal volume of 2 × SDS protein buffer (100 mM Tris-HCl pH 6.8, 200 mM dithiothreitol, 4% SDS, 0.2% bromophenol blue, 20% glycerol). Proteins were resolved by SDS-PAGE, transferred to polyvinylidine difluoride membrane (Polyscreen Dupont). Antibodies used for immunoblotting included, 0.5 μg/ml anti-cytochrome c 7H8.2C12 (Pharmingen), 0.1 μg/ml anti-HA rat antibody (Roche), 1.5 μg/ml anti-DRONC antibody,26 1 μg/ml anti-DRICE26 in 5% skim milk-PBST. The anti-DIAP1 and anti-DRONC (p24) antibodies were kindly provided by B Hay (Caltech). Anti-ARK rabbit antibody was generated in our laboratory and used as unpurified serum at 1 : 2000 dilution in 5% skim milk-PBS-T (PBS in 0.05% Tween-20). Secondary antibodies were conjugated with alkaline phosphatase and signals were detected by enhanced chemifluorescence (Amersham Biosciences).
Affinity capture of active DRONC
Cytosolic extracts (1 mg) from untreated or from cycloheximide-treated BG2 cells were incubated with 50 μM biotin-DEVD-CHO (BioMol International) or biotin-VAD-fluoromethylketone (Enzyme Systems) in binding buffer (50 mM HEPES pH 7.0, 50 mM NaCl, 0.1% CHAPS, 1 mM EDTA, 10% sucrose) at 27°C for 1 h. Biotinylated proteins were pulled down with streptavidin-conjugated sepharose (Amersham), washed in 10 volumes of binding buffer and bound proteins were eluted from the beads by boiling in SDS protein buffer. Where indicated, 50 μM zVAD was pre-incubated with extracts for 30 min at 27°C prior to addition of biotin-VAD. Samples were separated through 15% SDS-PAGE and analysed by immunoblotting with anti-DRONC or anti-DRICE antibodies.
Gel filtration chromatography
BG2 cells (1 × 108) were resuspended in buffer A containing 250 mM sucrose and lysed by homogenisation. Cell lysates were cleared by centrifugation at 100 000 g, 30 min at 4°C. Protein lysate (5 mg) or 1 μg of recombinant ARK and DRONC proteins were fractionated using FPLC protein purification system on a Superdex 200 chromatography column (Amersham Biosciences) at 4°C as previously described.26
Abbreviations
- CARD:
-
caspase recruitment domain
- IVT:
-
in vitro translated
- AMC:
-
7-amino-4-methylcoumarin
- AFC:
-
7-amino-4-trifluoromethyl coumarin
References
Kumar S . Caspase function in programmed cell death. Cell Death Differ 2007; 14: 32–43.
Boatright KM, Salvesen GS . Mechanisms of caspase activation. Curr Opin Cell Biol 2003; 15: 725–731.
Walker NP, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenz CR et al. Crystal structure of the cysteine protease interleukin-1 beta-converting enzyme: a (p20/p10)2 homodimer. Cell 1994; 78: 343–352.
Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA et al. Structure and mechanism of interleukin-1 beta converting enzyme. Nature 1994; 370: 270–275.
Rotonda J, Nicholson DW, Fazil KM, Gallant M, Gareau Y, Labelle M et al. The three-dimensional structure of apopain/CPP32, a key mediator of apoptosis. Nat Struct Biol 1996; 3: 619–625.
Fuentes-Prior P, Salvesen GS . The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J 2004; 384: 201–232.
Saikumar P, Mikhailova M, Pandeswara SL . Regulation of caspase-9 activity by differential binding to the apoptosome complex. Front Biosci 2007; 12: 3343–3354.
Bao Q, Shi Y . Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ 2007; 14: 56–65.
Timmer JC, Salvesen GS . Caspase substrates. Cell Death Differ 2007; 14: 66–72.
Wang X . The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15: 2922–2933.
Rodriguez J, Lazebnik Y . Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev 1999; 13: 3179–3184.
Zou H, Li Y, Liu X, Wang X . An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 1999; 274: 11549–11556.
Guerrero AD, Chen M, Wang J . Delineation of the caspase-9 signaling cascade. Apoptosis 2007 in press (doi:10.1007/s10495-007-0139-8).
Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS . Caspase-9 can be activated without proteolytic processing. J Biol Chem 1999; 274: 8359–8362.
Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW . Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 2002; 9: 423–432.
Pop C, Timmer J, Sperandio S, Salvesen GS . The apoptosome activates caspase-9 by dimerization. Mol Cell 2006; 22: 269–275.
Yin Q, Park HH, Chung JY, Lin SC, Lo YC, da Graca LS et al. Caspase-9 holoenzyme is a specific and optimal procaspase-3 processing machine. Mol Cell 2006; 22: 259–268.
Chao Y, Shiozaki EN, Srinivasula SM, Rigotti DJ, Fairman R, Shi Y . Engineering a dimeric caspase-9: a re-evaluation of the induced proximity model for caspase activation. PLoS Biol 2005; 3: e183.
Denault JB, Eckelman BP, Shin H, Pop C, Salvesen GS . Caspase 3 attenuates XIAP (X-linked inhibitor of apoptosis protein)-mediated inhibition of caspase 9. Biochem J 2007; 405: 11–19.
Bratton SB, Walker G, Srinivasula SM, Sun XM, Butterworth M, Alnemri ES et al. Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J 2001; 20: 998–1009.
Daish TJ, Mills K, Kumar S . Drosophila caspase DRONC is required for specific developmental cell death pathways and stress-induced apoptosis. Dev Cell 2004; 7: 909–915.
Chew SK, Akdemir F, Chen P, Lu WJ, Mills K, Daish T et al. The apical caspase dronc governs programmed and unprogrammed cell death in Drosophila. Dev Cell 2004; 7: 897–907.
Mills K, Daish T, Kumar S . The function of the Drosophila caspase DRONC in cell death and development. Cell Cycle 2005; 4: 744–746.
Waldhuber M, Emoto K, Petritsch C . The Drosophila caspase DRONC is required for metamorphosis and cell death in response to irradiation and developmental signals. Mech Dev 2005; 122: 914–927.
Xu D, Li Y, Arcaro M, Lackey M, Bergmann A . The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development 2005; 132: 2125–2134.
Dorstyn L, Read S, Cakouros D, Huh JR, Hay BA, Kumar S . The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J Cell Biol 2002; 156: 1089–1098.
Zimmermann KC, Ricci JE, Droin NM, Green DR . The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol 2002; 156: 1077–1087.
Mills K, Daish T, Harvey KF, Pfleger CM, Hariharan IK, Kumar S . The Drosophila melanogaster Apaf-1 homologue ARK is required for most, but not all, programmed cell death. J Cell Biol 2006; 172: 809–815.
Yu X, Wang L, Acehan D, Wang X, Akey CW . Three-dimensional structure of a double apoptosome formed by the Drosophila Apaf-1 related killer. J Mol Biol 2006; 355: 577–589.
Yan N, Huh JR, Schirf V, Demeler B, Hay BA, Shi Y . Structure and activation mechanism of the Drosophila initiator caspase Dronc. J Biol Chem 2006; 281: 8667–8674.
Hawkins CJ, Yoo SJ, Peterson EP, Wang SL, Vernooy SY, Hay BA . The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J Biol Chem 2000; 275: 27084–27093.
Muro I, Monser K, Clem RJ . Mechanism of Dronc activation in Drosophila cells. J Cell Sci 2004; 117: 5035–5041.
Meier P, Silke J, Leevers SJ, Evan GI . The Drosophila caspase DRONC is regulated by DIAP1. EMBO J 2000; 19: 598–611.
Muro I, Hay BA, Clem RJ . The Drosophila DIAP1 protein is required to prevent accumulation of a continuously generated, processed form of the apical caspase DRONC. J Biol Chem 2002; 277: 49644–49650.
Yoo SJ, Huh JR, Muro I, Yu H, Wang L, Wang SL et al. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat Cell Biol 2002; 4: 416–424.
Holley CL, Olson MR, Colon-Ramos DA, Kornbluth S . Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat Cell Biol 2002; 4: 439–444.
Muro I, Means JC, Clem RJ . Cleavage of the apoptosis inhibitor DIAP1 by the apical caspase DRONC in both normal and apoptotic Drosophila cells. J Biol Chem 2005; 280: 18683–18688.
Baliga BC, Read SH, Kumar S . The biochemical mechanism of caspase-2 activation. Cell Death Differ 2004; 11: 1234–1241.
Dorstyn L, Colussi PA, Quinn LM, Richardson H, Kumar S . DRONC, an ecdysone-inducible Drosophila caspase. Proc Natl Acad Sci USA 1999; 96: 4307–4312.
Yan N, Wu JW, Chai J, Li W, Shi Y . Molecular mechanisms of DrICE inhibition by DIAP1 and removal of inhibition by Reaper, Hid and Grim. Nat Struct Mol Biol 2004; 11: 420–428.
Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS . Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci USA 2001; 98: 14250–14255.
Acknowledgements
This study was supported by the National Health and Medical Research Council of Australia (SK) and a Royal Adelaide Hospital Florey Research Fellowship (LD).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by E Baehrecke
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Dorstyn, L., Kumar, S. A biochemical analysis of the activation of the Drosophila caspase DRONC. Cell Death Differ 15, 461–470 (2008). https://doi.org/10.1038/sj.cdd.4402288
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402288
Keywords
This article is cited by
-
Accurate elimination of superfluous attachment cells is critical for the construction of functional multicellular proprioceptors in Drosophila
Cell Death & Differentiation (2019)
-
New insights into apoptosome structure and function
Cell Death & Differentiation (2018)
-
Hedgehog signalling is required for cell survival in Drosophila wing pouch cells
Scientific Reports (2017)
-
The deubiquitinating enzyme DUBAI stabilizes DIAP1 to suppress Drosophila apoptosis
Cell Death & Differentiation (2014)
-
The role of IAP antagonist proteins in the core apoptosis pathway of the mosquito disease vector Aedes aegypti
Apoptosis (2011)

{kind=link}
{kind=link}