
Abstract
Smac/Diablo and HtrA2/Omi promote apoptosis by binding to and antagonizing IAP proteins, including the ‘X chromosome-linked inhibitor of apoptosis’ (XIAP). Here we show that caspase-mediated proteolysis of a limited subset of cell death substrates exposes functional Smac/Diablo-like N-termini after cleavage, which are able to bind to and antagonize XIAP. We propose that this mechanism may establish a feedforward sensitization of the apoptotic pathway and contribute to the functional redundancy of IAP antagonism. In addition, this may be particularly relevant in Alzheimer's disease since the caspase-generated C31 peptide, an established cytotoxin, acquires Smac/Diablo-like properties after apoptotic processing.
Similar content being viewed by others


Introduction
XIAP performs its antiapoptotic activity by binding and inhibiting caspase-3, -7 and -9.1 Two proapoptotic mitochondrial proteins, Smac/Diablo and HtrA2/Omi, interact with and antagonize XIAP, thereby restoring caspase activity and promoting cell death.2,3,4,5,6,7,8 Both proteins are synthesized as larger precursors that are proteolytically processed during import into mitochondria. Processing unmasks residues that are essential for XIAP binding, including a critical N-terminal Ala residue, but release from mitochondria concomitant with cytochrome c is also required for accessibility to cytoplasmic XIAP. We reasoned that nonmitochondrial caspase substrates could perform a similar function if the N-terminus of the scissile bond contained a Smac/Diablo-like motif. This would obviate the need for mitochondrial perturbation, for example in some modes of ‘death receptor’ triggering, and also amplify the apoptotic response by a feedforward mechanism. A survey of 92 established cleavage sites within caspase substrates identified nine that would produce the critical N-terminal Ala (Figure 1a).9,10,11 We examined two of these in detail (Figure 1b):12,13 XIAP itself, since it could function as its own antagonist after caspase proteolysis in human cells, and the amyloid-β precursor protein (APP), since the caspase-generated C31 peptide has been shown to promote apoptotic death by a hitherto undefined mechanism.14

(a) A survey of 92 proteins that have sufficient criteria for being authentic caspase substrates during apoptotic death (see Nicholson,9 Roy and Nicholson10 and Fischer et al11 for details and further references) yields nine that would produce N-terminal Ala residues at the scissile bond (indicated by dashed vertical line). (b) Caspase-mediated cleavage of human XIAP and APP yield Ala N-termini with sequence similarity to established IAP antagonists. Synthetic peptides made to mimic these termini, and used in the experiments described below, are indicated in blue. The caspase cleavage site as well as the N-terminal Ala are unique to human XIAP (not present in rat, mouse or bovine sequences), whereas both are conserved in APP from most species (including mouse, rat, guinea-pig and others)
Experimental procedures
Equipment and reagents
The Biacore 1000, CM5 and SA sensor chips, N-hydroxysuccinimide (NHS), N-ethyl-N′-(3-diethylaminopropyl) carbodiimide hydrochloride (EDC) and 1 M ethanolamine (pH 8.5) were purchased from Biacore Inc. (Piscataway, NJ, USA). Synthetic peptides were purchased from Synprep Corporation (Dublin, CA, USA).
cDNA cloning, expression and purification of BIR3 (XIAP)
A cDNA encoding residues 244–497 of human XIAP (containing the BIR3 domain) plus a six histidine tag was created by PCR using the cDNA for XIAP (UMP 8411) as a template and the N-terminal primer XIAP-N244 (5′GCG TCT AGA CTC GAG CAT ATG GTG AGT TCT GAT AGG AAT TTC3′) and the C-terminal primer XIAP-C6HIS (5′GCG TCT AGA CTC GAG CAT ATG TTA ATG ATG ATG ATG ATG ATG AGA CAT AAA AAT TTT TTG CTT GAA AG3′). The resulting PCR fragment was cloned via the NdeI restriction site into the procaryotic expression vector pET11c (Novagen), generating the plasmid-DNA UMP 8684, which was confirmed by sequencing. The encoded BIR3 polypeptide was overexpressed in the E. coli strain Bl21-Codon Plus (DE3) (Novagen), and insoluble protein was recovered in purified inclusion bodies. The inclusion body BIR3 protein was dissolved in guanidine HCl (GuHCl) buffer (6 M GuHCl, 25 mM Na2HPO4 (pH 8.0), 5 mM DTT, 1 μM Zn(OAc)2), and then renatured by dialysis against 4 l of renaturing buffer RB (25 mM Na2HPO4 (pH 7.0), 5% (w/v) glycerol, 0.01% (v/v) NP-40, 1 mM DTT, 50 μM Zn(OAc)2) containing progressively decreasing concentrations of GuHCl. Dialysis was performed at 4°C using the following steps: 2 h 30 min RB+3 M GuHCl, 2 h 30 min RB+1 M GuHCl, 2 h 30 min RB+0.4 M GuHCl, 2 h 30 min RB+0.2 M GuHCl, o/n RB+0.1 M GuHCl, three times 2 h 30 min RB and finally o/n RB. The dialysed BIR3 solution was clarified by centrifugation at 3000 × g for 10 min at 4°C, followed by a second centrifugation at 30 000 × g for 15 min. The protein concentration in the resulting supernatant was determined by densitometric scanning of Coomassie blue-stained gels and by the method of Bradford. The protein solution was aliquoted, quick-frozen and stored at −80°C. Protein used for the surface plasmon resonance experiments was dialysed overnight against phosphate-buffered saline (PBS) plus 50 μM Zn(OAc)2.
Surface plasmon resonance
All surface plasmon resonance experiments were performed with an upgraded BIACORE 1000 at 25°C. The running buffer was HBS (20 mM HEPES (pH 7.4), 150 mM NaCl, 3.4 mM EDTA, 0.05% (v/v) Tween20) and the protein and peptide dilutions were done in HBS with 50 μM Zn(OAc)2.
Kinetic assay of the Smac-20 interaction with BIR3
Smac-20 interaction with BIR3 was performed on an SA sensorchip. The sensorchip surfaces were activated by three pulses (5 μl each) of 1 M NaCl and 50 mM NaOH. After baseline stabilization, Smac-20 was coupled to the surface through biotin–streptavidin interaction by injecting the biotinylated Smac-20 peptide (1 nM in HBS) until the desired amount was coupled (ca. 10 RU). The subsequent kinetic experiments were carried out at a flow rate of 20 μl/min. Different concentrations of BIR3 (from 46.7 to 1250 nM) were randomly injected over a Smac-20 surface as well as over a mock surface (no Smac-20) for 180 s, followed by buffer injection for 180 s. Regeneration of the sensor chip for subsequent injections was accomplished by two pulses of a solution of 50 mM n-octyl glucopyranoside, 10 mM HCl (25 μl each at 100 μl/min), followed by EXTRACLEAN and RINSE procedures. All experiments were done in duplicate. The curves were then prepared using the ‘double-referencing’ method and globally fit using nonlinear least squares analysis and numerical integration of the differential rate equations using the SPRevolution© software package. All the sensorgrams from injections of BIR3 concentrations over the Smac-20 surface were then globally analyzed using several different kinetic models that are available in the SPRevolution© software package.
Competition experiments
For the competition experiments, the flow rate was 5 μl/min. Smac-20 peptide was immobilized on CM5 sensorchips by the standard amine-coupling procedure. Mock surfaces were also generated using the same procedure by replacing the protein solution with running buffer. The competition experiments were run as follows: 1 μM of BIR3 or 1 μM of BIR3, preincubated for at least 1 h at room temperature with different concentrations of the competing peptides, was injected (15 μl) over the Smac-20 and mock surfaces. The amount of BIR3 still bound to the surface 5 s after the end of the injection on the mock surface was subtracted from the corresponding value from the Smac-20 surface. The quantity of BIR3 bound 5 s after the end of the injection was expressed as a percentage of BIR3 binding in the absence of competing peptide and plotted against the concentration of the competing peptide. After each injection, surface regeneration was accomplished by injecting 5 μl of 120 mM HCl solution. All the experiments were done at least in duplicate.
Preparation of cytoplasmic S-100 cell extract
Frozen Jurkat cells (equivalent to 5.2 × 109 cells) were resuspended in 5 volumes of buffer A (20 mM HEPES-KOH (pH 7.5), 10 mM KCl, 2 mM Na-EDTA, 250 mM sucrose, 1 mM DTT) containing 0.1 mM PMSF and 1% (v/v) Sigma P-8340 Protease-Inhibitor-Mix. Following incubation on ice for 15 min, the cells were disrupted using a Dounce homogenizator (15 strokes, Wheaton 15 ml, Pestle B) and then centrifuged at 1000 × g for 10 min at 4°C to remove nuclei and cell debris. The resulting supernatant was centrifuged at 13 000 × g for 10 min at 4°C, followed by a third centrifugation step at 100 000 × g for 1 h at 4°C. The resulting supernatant was aliquoted, frozen on dry ice and stored at −80°C.
Cytochrome c-dependent caspase activation assay
The cytochrome c-dependent activation assay was performed in a 20 μl volume of reaction buffer A (20 mM HEPES-KOH (pH 7.5), 10 mM KCl, 2 mM Na-EDTA, 250 mM sucrose, 1 mM DTT). Protein (50 μg) from the S-100 cell extract was mixed into 40 nM of purified, recombinant BIR3 (XIAP) and variable amounts of peptides or vehicle. The reaction was then started by the addition of 1 μg of horse heart cytochrome c, 1.25 mM dATP and 2.5 mM MgCl2 and incubated for 1 h at 30°C. Cytochrome c-dependent activation of caspase-9 was monitored via caspase-3 activity, which was measured by fluorometry as release of AMC from Ac-DEVD-AMC. Following the activation reaction, 180 μl of caspase-3 substrate buffer was added (50 mM HEPES-KOH (pH 7.0), 2 mM Na-EDTA, 0.1% (w/v) CHAPS, 10% (w/v) sucrose supplemented with the substrate Ac-DEVD-AMC and DTT) to obtain final concentrations of 10 μM (Ac-DEVD-AMC) and 5 mM (DTT). Using an excitation wavelength of 380 nm and an emission wavelength of 460 nm, the release of free AMC was monitored continuously for 60 min in 1-min intervals and expressed as arbitrary fluorescence units per min. The DEVDase activity of the mock-treated sample without BIR3 inhibition was set to 100% and used as reference for the other samples.
Results and discussion
For these studies, we chose to study the antagonism of the BIR3 domain of XIAP, which works at the apical point of the caspase cascade by preferentially inhibiting caspase-9.12 The binding constants for Smac/Diablo antagonism of BIR2 have previously been established by surface plasmon resonance (Kd=35 nM).15 Smac-20, a synthetic peptide containing the N-terminal 20 residues of mature Smac and able to bind BIR2,16 bound to BIR3 with a Kd value of 64 nM, consistent with it also being a high-affinity antagonist of BIR3 (Figure 2a). Using the interaction of Smac-20 with BIR3 as a positive control, the entire proapoptotic caspase-generated C31 peptide of APP (APP-AA), or a 12-residue XIAP peptide corresponding to the neo-N terminus generated by caspase proteolysis (XIAP-AV), was tested for BIR3 binding at the same site by competition surface plasmon resonance (Figure 2b). All three Ala-terminal peptides were able to compete for the Smac/Diablo-binding site on BIR3, whereas control peptides with altered N-termini did not. The rank order of affinity was Smac-20>APP-AA>XIAP-AV. These data indicate that caspase-generated Ala termini within substrates, such as APP and XIAP, are biochemically able to bind to the Smac/Diablo groove in BIR3.

(a) Surface plasmon resonance kinetic analysis of the interaction of Smac-20 with the BIR3 of XIAP. A C-terminal biotinylated Smac-20 peptide (corresponding to the first 20 residues of mature mitochondrial Smac/Diablo) was immobilized on a Biacore SA chip and the indicated concentrations of recombinant BIR3 (residues 244–497 of human XIAP, containing the C-terminal ring-zinc-finger and a six-His epitope tag) were injected over the ligand surface for 180 s. A Kd of 64 nM was calculated from the resulting response curves. (b) Peptides corresponding to caspase-generated fragments of XIAP and APP (XIAP-AV 12 mer and APP-AA 31 mer, respectively) compete for binding to the Smac/Diablo site on BIR3. A nonbiotinylated Smac-20 peptide was immobilized by amine coupling to a Biacore CM5 sensorchip. Solutions of the indicated peptide concentrations were preincubated with 2.5 μM recombinant BIR3 prior to injection onto the immobilized Smac-20 chip. Competition curves were constructed from the resulting sensorgrams. Two control peptides with altered N-termini (MetGly N-terminus for APP instead of AlaAla (APP-MG), Met N-terminus for XIAP instead of Ala (XIAP-MV)) were tested at the highest concentrations used without effect
We next wanted to determine whether these same peptides could functionally antagonize BIR3 inhibition of caspase activation. For these experiments, we used the cytochrome c-responsive in vitro activation system17 used previously to define Smac/Diablo function.2,3,4 Isolated cell cytosols were activated with cytochrome c plus dATP. This results in the formation of an active apoptosome, caspase-9 activation and then caspase-3 activation, which is measured in a continuous fluorometric assay using the caspase-3 substrate, Ac-DEVD-AMC.18 The recombinant BIR3 domain of XIAP blocked caspase activation in this system and this blockade was entirely prevented by the presence of ⩾50 μM of Smac-20, the positive control synthetic peptide (Figure 3a). Similarly, both the APP-C31 peptide and the XIAP-AV peptide (Figure 3b and c, respectively) were able to antagonize the BIR-3-mediated attenuation of cytochrome c-stimulated caspase activation, but the corresponding control peptides were not. The rank order of efficacy was entirely consistent with the biophysical interaction studies. Furthermore, in the absence of exogenously added BIR3, apoptosome activation was enhanced approximately 20% by the presence of Smac-20, APP-C31 or XIAP-AV (but not their corresponding control peptides), presumably owing to antagonism of endogenous IAPs already present in the Jurkat cell cytosol fractions (Figure 4). In this system (absence of exogenous BIR3), maximum stimulation occurred with all three peptides at 12.5 μM.

Peptides corresponding to mature Smac/Diablo (a) or to the caspase-generated fragments of APP (b) and XIAP (c) antagonize IAP-mediated inhibition of caspase activation. The apoptosome/caspase-9/caspase-3 pathway was stimulated in naïve S100 Jurkat cell cytosols by the addition of cytochrome c (50 ng/μl) and dATP/MgCl2 (1.25 mM/2.5 mM). After a 1 h incubation, caspase-3 activity was measured by continuous fluorometry using 10 μM Ac-DEVD-AMC. All samples contained 40 nM recombinant BIR3 (described above), except reference samples used to establish the 100% ‘noninhibited’ activity. The indicated active peptides (solid bars) and negative control peptides (open bars) were included in the incubations at the concentrations indicated

Peptides corresponding to mature Smac/Diablo (a) or to the caspase-generated fragments of APP (b) and XIAP (c) antagonize endogenous IAP-mediated inhibition of caspase activation and accelerate caspase activation. The apoptosome/caspase-9/caspase-3 pathway was stimulated in naïve S100 Jurkat cell cytosols exactly as described for Figure 3, except that recombinant BIR3 was not included in the incubation mixtures. The indicated active peptides (solid bars) and negative control peptides (open bars) were included in the incubations at the concentrations indicated
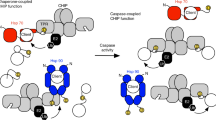
Collectively, these data demonstrate that in addition to the two mitochondrial proteins previously demonstrated to antagonize IAP function, and the caspase-9 linker peptide which baits XIAP,19 caspase substrates may themselves function to promote amplification of the caspase cascade by participating in the antagonism of IAP function. This could promote a feedforward sensitization of the apoptotic pathway (Figure 5) and may be of particular relevance in Alzheimer's disease, where the caspase-generated C31 peptide of APP has been shown to promote apoptotic death. These findings also predict that IAP antagonism is a collective event, and that the absence of an overt phenotype in Smac/Diablo-deficient mice20 is expected given the redundancy that occurs as a consequence of caspase cleavage products having IAP antagonistic properties.

Feedforward amplification model for apoptosis sensitization by caspase-substrate neo-termini. IAP proteins, such as XIAP, help prevent inappropriate apoptotic suicide by direct inhibition of activator and effector caspases (C9, C3, C7; caspase-9, -3 and -7, respectively). Apoptotic triggering through the ‘intrinsic’ mitochondrial pathway is initiated by the release of mitochondrial intermembrane space constituents including cytochrome c (circled ‘c’), which is a cofactor for APAF-1 oligomerization and activation of caspase-9, and Smac/Diablo plus HtrA2/Omi, which antagonize the IAP-mediated blockade on caspase activity through P1′ Ala N-termini. IAPs are also antagonized by the pro-caspase-9 linker peptide which is exposed following caspase-9 proteolytic maturation (not indicated). A limited subset of caspase cell death substrates present Smac/Diablo-like P1′ Ala N-termini when cleaved by the effector caspases – caspase-3 and -7 (e.g. XIAP and APP). These neo-termini also participate in IAP antagonism, thus contributing to the unopposed activation of the caspase pathway
Abbreviations
- caspase:
-
cysteinyl aspartate-specific proteinase
- IAP:
-
inhibitor of apoptosis proteins
- XIAP:
-
X-linked IAP
- BIR:
-
baculovirus IAP repeat
- Smac:
-
second mitochondria-derived activator of caspase
- Diablo:
-
direct IAP-binding protein with low pI
- APP:
-
amyloid-β precursor protein
References
Deveraux QL, Takahashi R, Salvesen GS and Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300–304
Du C, Fang M, Li Y, Li L and Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ and Vaux DL (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53
Srinivasula SM, Datta P, Fan XP, Fernandes-Alnemri T, Huang Z and Alnemri ES (2000) Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J. Biol. Chem. 275: 36152–36157
Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K and Takahashi R (2000) A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 8: 613–621
Hegde R, Srinisasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri R and Alnemri ES (2002) Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein–caspase interaction. J. Biol. Chem. 277: 432–438
Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C and Downward J (2002) The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J. Biol. Chem. 277: 439–444
Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz RL, Simpson RJ and Vaux DL (2002) HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem. 277: 445–454
Nicholson DW (1999) Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 6: 1028–1042
Roy S and Nicholson DW (2000) Criteria for identifying authentic caspase substrates during apoptosis. Methods Enzymol. 322: 110–125
Fischer U, Jänicke RU and Schulze-Osthoff K (2003) Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 10: 76–100
Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS and Reed JC (1999) Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 18: 5242–5251
Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LHT, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S and Nicholson DW (1999) Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell 97: 395–406
Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH and Bredesen DE (2000) A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat. Med. 6: 397–404
Huang Y, Park YC, Rich RL, Segal D, Myszka DG and Wu H (2001) Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 104: 781–790
Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC and Fesik SW (2000) Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 408: 1004–1008
Liu X, Kim CN, Yang J, Jemmerson R and Wang X (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Nature 86: 147–157
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju AM, Smulson ME, Yamin T-T, Yu VL and Miller DK (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376: 37–43
Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y and Alnemri ES (2001) A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410: 112–116
Okada H, Suh W-K, Jin J, Woo M, Du C, Elia A, Duncan GS, Wakeham A, Itie A, Lowe SW, Wang X and Mak TW (2002) Generation and characterization of Smac/DIABLO-deficient mice. Mol. Cell Biol. 22: 3509–3517
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Hell, K., Saleh, M., Crescenzo, G. et al. Substrate cleavage by caspases generates protein fragments with Smac/Diablo-like activities. Cell Death Differ 10, 1234–1239 (2003). https://doi.org/10.1038/sj.cdd.4401298
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401298
Keywords
This article is cited by
-
More alive than dead: non-apoptotic roles for caspases in neuronal development, plasticity and disease
Cell Death & Differentiation (2017)
-
Apoptosis induction of U937 human leukemia cells by diallyl trisulfide induces through generation of reactive oxygen species
Journal of Biomedical Science (2012)
-
The mitochondrial serine protease HtrA2/Omi: an overview
Cell Death & Differentiation (2008)
-
Anti-myeloma effect of homoharringtonine with concomitant targeting of the myeloma-promoting molecules, Mcl-1, XIAP, and β-catenin
International Journal of Hematology (2008)
-
Caspase substrates
Cell Death & Differentiation (2007)
