
Abstract
We have documented previously somatic mutations of STK11/LKB11, the gene responsible for Peutz-Jeghers syndrome (PJS), in a small proportion of sporadic pancreatic adenocarcinomas, intraductal papillary mucinous neoplasms (IPMNs), and biliary adenocarcinomas. In this report, we characterize the expression of Stk11, the protein product of the STK11 gene, in a larger series of pancreatic and biliary neoplasms. First, the specificity of the Stk11 antibody was established in 23 neoplasms (22 IPMNs and 1 biliary adenocarcinoma) with known STK11 gene status. Complete absence of labeling was seen in the neoplastic cells of 3 of the 3 (100%) cases with previously documented biallelic inactivation of the STK11 gene, whereas 16 of the 20 (80%) IPMNs, presumably with at least one wild-type STK11 gene, retained Stk11 expression in the neoplastic cells. The marked decrease or absence of Stk11 expression in four neoplasms with wild-type STK11 suggests that additional mechanisms may account for the lack of Stk11 expression. Subsequently, to further evaluate Stk11 expression in pancreatic and biliary neoplasms, tissue microarrays (TMAs) were constructed from a series of nearly 100 ductal adenocarcinomas and biliary neoplasms. Stk11 expression was lost in 4 of the 56 (7%) pancreatic adenocarcinomas and 1 of the 38 (2.6%) biliary cancers by immunohistochemistry; the absence of labeling was confirmed by repeated immunohistochemical labeling of complete tissue sections for the same cases. Thus, Stk11 expression is abrogated in a small proportion of pancreatic and biliary neoplasms. The inactivation of Stk11 in 27% (6/22) of IPMNs versus 7% (4/56) of pancreatic adenocarcinomas suggests genetic disparities in the pathogenesis of these closely related neoplasms. Immunohistochemical analysis for Stk11 expression may be a valid surrogate for genetic analysis of STK11 gene mutations in cancers.
Similar content being viewed by others

INTRODUCTION
PJS is an autosomal-dominant disorder characterized by hamartomatous polyps in the gastrointestinal tract and pigmented macules of the lips and buccal mucosa (1). PJS has also been associated with an increased risk of developing cancers (2) including gastrointestinal, gynecologic, lung, breast, thyroid, basal cell, prostate, and pancreatic cancers (3, 4, 5). PJS is caused by germline mutations of the STK11 gene (also known as LKB1) on distal chromosome 19p, which encodes a novel serine/threonine kinase (6, 7).
We have previously demonstrated biallelic inactivation of the STK11 gene in a pancreatic cancer that developed in a patient with PJS (8); similarly, somatic STK11 gene mutations have been demonstrated in a small proportion of sporadic pancreatic and biliary cancers (8, 9). Somatic STK11 gene mutations have also been reported in lung adenocarcinomas, malignant melanomas, and ovarian tumors (10, 11, 12), all supporting the role of STK11 as a tumor-suppressor gene.
Recent studies have indicated that STK11 mediates cell cycle arrest through induction of the cyclin-dependent kinase inhibitor p21WAF1 (13), through a p53-dependent process. The Stk11 protein also interacts with brahma-related gene-1 (BRG1), an ATPase that is associated with SWI/SNF chromatin-remodeling complexes. Exogenous expression of brg1 is able to induce cell cycle arrest and senescence in a retinoblastoma-dependent fashion (14). These studies suggest that the tumor-suppressor function of STK11 lies within its ability to affect the cell cycle proliferation.
In this report we describe for the first time an antibody that specifically labels the Stk11 protein. To determine whether loss of Stk11 protein expression is a predictor of genetic mutation of STK11, we performed immunohistochemical analysis on a large series of pancreatic and biliary neoplasms. We demonstrate that immunohistochemical labeling for Stk11 expression is a reliable surrogate for genetic analysis in archival tissues.
Furthermore, our results confirm the previously reported role of STK11 gene as a tumor-suppressor gene in pancreatic and biliary neoplasms. We also have uncovered a larger role for STK11 in IPMN than in ductal pancreatic adenocarcinoma, making STK11 the first tumor-suppressor gene identified whose function may be more prevalently abrogated in IPMNs than in pancreatic ductal adenocarcinomas.
MATERIAL AND METHODS
Selection of Previously Characterized Pancreatobiliary Neoplasms
We have previously characterized the status of the STK11 gene by mutation analysis in a series of 22 IPMNs and 1 biliary adenocarcinoma (8, 9). This series of 23 cases included 3 cases with bi-allelic loss of STK11 gene function. We designated these cases as our “training set” for optimization of the Stk11 antibody. The archival blocks on these 23 cases were retrieved from the surgical pathology archives of the Johns Hopkins Hospital, and presence of adequate neoplastic cellularity was confirmed on selected blocks. Five-micrometer tissue sections were then prepared on ChemMate (DAKO, Carpinteria, CA) -charged slides for Stk11 immunohistochemistry.
Preparation of Pancreatobiliary Cancer Tissue Microarrays
Formalin-fixed paraffin-embedded archival tissue samples from 68 sporadic pancreatic adenocarcinomas and 38 sporadic biliary neoplasms (13 gallbladder adenocarcinomas, 15 distal bile duct adenocarcinomas, and 10 intrahepatic cholangiocarcinomas) were retrieved from the surgical pathology archives of the Johns Hopkins Hospital. These cases have not previously been characterized with regards to the mutational status of their STK11 gene. Hematoxylin and eosin stained sections from these cases were reviewed by three pathologists (AM, PA, and RHH) to confirm the diagnosis and the presence of neoplastic cells on selected tissue blocks. Subsequently, representative areas containing neoplastic cells were circled on the glass slides and used as a template. Tissue microarrays were constructed using a manual Tissue Puncher/Arrayer (Beecher Instruments, Silver Spring, MD), as previously described (15). For each cancer, four 1.4-mm cores were carefully selected from tumor-rich areas of the donor block to ensure that adequate numbers of neoplastic cells were represented in the recipient block. Two serial sections were cut from the recipient TMA block, one of which was stained with hematoxylin and eosin as a reference, and the second for Stk11 immunohistochemistry. Of the 68 pancreatic adenocarcinomas selected, 12 (18%) cases were discarded because of loss of multiple tissue cores in the Stk11-stained section or because of inadequate representation of neoplastic cells on the retained cores. The remaining 56 (82%) pancreatic adenocarcinomas were chosen for the final immunohistochemical analysis. Thirty-eight of 38 biliary neoplasms had adequate neoplastic cellularity on the Stk11-stained microarray and were selected for the final immunohistochemical analysis. As an additional measure of stringency to avoid false negatives (i.e., mistakenly designating a case as Stk11 negative because of tumor heterogeneity), full sections of tumor were restained in those cases that demonstrated loss of Stk11 labeling in the TMA cores to confirm the absence of labeling in the neoplastic cells.
Immunohistochemistry
Both regular tissue sections and TMA sections were labeled using identical protocols. Unstained 5-μm sections were cut from the respective paraffin blocks and deparaffinized by routine techniques. The slides were treated with sodium citrate buffer (diluted to 1 × from 10 × heat-induced epitope retrieval buffer; Ventana-Bio Tek Solutions, Tucson, AZ) and steamed for 20 minutes at 90° C. After cooling for 5 minutes, the slides were labeled with polyclonal antibody to Stk11 (dilution of 1:10; Cell Signaling, Beverly, MA) using the DAKO autostainer, followed by addition of biotinylated secondary antibodies, avidin-biotin complex, and 3,3′-diaminobenzidine (LSAB Plus kit; DAKO). Sections were counterstained with Giles hematoxylin.
Three authors of this study (AM, RHH, and PA) independently evaluated the immunohistochemical labeling of the carcinomas. Each evaluator was unaware of the STK11 gene status of each case. Normal pancreatic ducts, islets of Langerhans, and lymphocytes, which demonstrate moderate to strong labeling with Stk11 protein, served as positive internal controls in each of the sections. Labeling in the tumors was scored as positive, markedly decreased, or negative. “Positive” labeling was defined as strong and uniform expression of Stk11 in >10% of neoplastic cells; however, in virtually all positive cases, ≥75% of tumor cells were labeled with Stk11, and only patchy negativity was noted. Cases were regarded as negative when no expression was seen in the nuclear or cytoplasmic compartments of all tumor cells. Cases were defined as markedly decreased when there was faint labeling compared with the case of normal tissues in <10% of neoplastic cells.
RESULTS
Stk11 Expression in Normal Pancreas
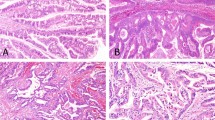
The antibody to Stk11 was first tested by immunohistochemical labeling of the nonneoplastic tissues of pancreas (Fig. 1a), where Stk11 expression was primarily restricted to the ductal epithelium. Cytoplasmic labeling with a membranous accentuation was observed in the ductal epithelial cells. Acinar, stromal, and islet cells were not labeled. The labeling was specific, as no positive labeling was observed when the primary antibody was omitted.

A, Stk11 expression is restricted to the ductal epithelium of non-neoplastic pancreas (black arrow). Non-neoplastic pancreatic ducts demonstrate cytoplasmic labeling with a membranous accentuation, whereas the islet cells and stromal cells exhibit a complete absence of labeling (white arrow). B, invasive component of an IPMN demonstrating loss of Stk11 labeling (white arrow), whereas adjacent non-neoplastic ducts are positive (black arrow). C, noninvasive component of an IPMN demonstrating loss of Stk11 (white arrow), whereas non-neoplastic ducts are positive (black arrow). D, biliary adenocarcinoma demonstrating loss of Stk11 labeling (white arrow), whereas non-neoplastic ducts show intact expression (black arrow).
Immunolabeling of 23 Previously Genetically Characterized Pancreatobiliary Neoplasms
To determine the specificity of the Stk11 antibody, we first characterized 22 IPMNs and 1 biliary adenocarcinoma with known genetic status at the STK11 locus (8, 9). Each case was scored by the three reviewers without the knowledge of its STK11 gene status.
Histologically, all IPMNs were characterized by having tall, columnar, mucin-containing neoplastic epithelium with or without papillary proliferations. The intraductal components of the 22 IPMNs were classified as adenoma in 1 (4%), borderline in 5 (23%), and carcinoma in situ in 16 (73%). Twelve (55%) of the IPMN were associated with infiltrating adenocarcinomas. Among the 22 IPMN cases, 2 were from patients harboring germline STK11 mutations, with somatic inactivation of the second allele in the IPMN (STK11 LOH + mutation). Four additional cases had loss of heterozygosity at the STK11 locus but did not harbor mutations in the STK11 gene (STK11 LOH) (9).
Stk11 labeling was absent or markedly decreased in 6 (27%) of the 22 IPMNs. In all six cases, the normal ducts strongly expressed Stk11, whereas the neoplastic cells did not (Fig. 1B–C). Notably, both IPMNs with biallelic inactivation of the STK11 gene (STK11 LOH + mutation) did not label with the antibody to the Stk11 protein, whereas adjacent normal ducts in the same case labeled diffusely. Both cases were diagnosed as carcinoma in situ. The remaining four IPMNs demonstrating none or markedly decreased labeling had previously been described as “wild type” for STK11 (no LOH at the STK11 locus) (Table 1). The immunohistochemical labeling of these 4 cases was repeated twice to confirm the absence of labeling in the neoplastic cells; antigenicity of tissue was confirmed by the presence of intact diffuse staining in adjacent nonneoplastic ductal epithelium. These four cases were classified as carcinoma in situ (n = 2), borderline (n = 1), and adenoma (n = 1), and there was no variability in the degree of atypia within the tumor block examined. Nevertheless, loss of Stk11 expression is apparently not restricted to invasive or highly atypical (i.e., carcinoma in situ) components only, but rather, the loss of expression in the one adenoma suggests that this abnormality may be an “early” event in IPMN progression. It is presumed that the conventional lack of stk11 expression in these four cases was caused by mechanism other than genetic mutation (STK11 LOH + mutation). Probable mechanisms are discussed later. Finally, positive Stk11 labeling was seen in all four IPMNs previously described with LOH at 19p13.3 (STK11 LOH) with no mutation in the remaining allele (Table 1).
We also labeled a biliary adenocarcinoma case with known homozygous deletion at the STK11 locus (8). Nonneoplastic biliary epithelium expressed Stk11, but no expression was seen in the neoplastic cells (Fig. 1D). Overall, we found that 16 of the 20 (80%) primary cancers with at least one demonstrable wild-type STK11 allele expressed Stk11 protein, whereas 3 of the 3 (100%) primary cancers with biallelic inactivated STK11 gene did not.
Analysis of the Stk11 Expression in TMAs of Pancreatobiliary Cancers
Initially, 6 of 56 pancreatic adenocarcinomas on the pancreatic TMA demonstrated lack of Stk11 expression. To exclude false positives for loss of Stk11 labeling, immunohistochemistry on these six samples was repeated on whole-tissue sections prepared from two different blocks. Four of the six adenocarcinomas were persistently negative for Stk11 expression on repeat immunohistochemical assays (data not shown), whereas in two cases, loss of labeling was focal. In summary, 4 of the 56 (7%) pancreatic adenocarcinoma cases demonstrated a lack of Stk11 expression (Table 2). All four cases were classified as adenocarcinoma and did not show special pathological findings.
The biliary TMA contained 15 distal bile duct carcinomas, 13 gall bladder adenocarcinomas, 10 intrahepatic cholangiocarcinomas, and 5 normal gallbladders. Initially, 4 adenocarcinomas demonstrated absent or markedly decreased Stk11 expression on the TMA cores. The labeling of these four cases was repeated with whole-tissue sections made from two different blocks. One intrahepatic cholangiocarcinoma demonstrated persistent lack of Stk11 labeling (data not shown), whereas in the other three cases, the loss of labeling was focal. Therefore, 1 of the 38 (2.6%) biliary adenocarcinomas showed diffuse loss of Stk11 expression (Table 2).
DISCUSSION
Immunohistochemical analysis of protein expression provides several distinct advantages over mutational analysis in archival tissue specimens. Mutation analysis of tumor-suppressor genes such as STK11 can be a challenging task, especially in pancreatic adenocarcinomas, which evoke an intense desmoplastic response. Examining all exons of the STK11 gene by sequencing is usually not a cost-effective proposition. It is nearly impossible to sequence all exons of the STK11 gene with the limited amount of DNA from microdissected neoplastic cells of a pancreatic adenocarcinoma. On the contrary, a re1iable immunological assay allows a large number of archived, formalin-fixed tissues to be examined in a high-throughput fashion.
In this report, we describe an antibody that is specific for the protein product of the STK11 gene and characterize its distribution in pancreatobiliary neoplasms. We included a subset of 23 cancers with previously known STK11 gene status as controls for this newly described antibody. Notably, three of the three cancers (two IPMNs, one biliary adenocarcinoma) with biallelic inactivation of the STK11 gene demonstrated complete absence of labeling in the neoplastic cells, whereas adjacent nonneoplastic epithelium was diffusely positive. In addition, 16 of the 20 (80%) cancers, presumably with at least one wild-type STK11 allele, demonstrated intact Stk11 expression in the neoplastic cells. Interestingly, the 4 of the 20 IPMNs with previously described wild-type STK11 alleles demonstrated absence of or markedly decreased Stk11 labeling in the neoplastic cells, despite multiple attempts at staining. Tissue antigenicity in all four cases was confirmed by positive internal controls (adjacent non-neoplastic ducts were diffusely positive). There are several possible explanations that can account for the loss of stk11 expression in neoplasms with presumed intact STK11 gene. In the previous study by Sato et al., only coding sequence mutations were analyzed only in the cases that exhibited LOH at the STK11 locus. Therefore, these four cases were never analyzed by sequencing because they did not exhibit LOH at the STK11 locus (9). It is possible that these four cases harbored a small intragenic mutation on each of their two alleles, a less frequent biallelic inactivation that has been shown to occur in other tumor-suppressor genes (16). It is also possible that these cases may harbor promoter, intronic, or splice junction mutations (17, 18). In addition, absent or decreased expression of Stk11 may be caused by means other than genetic mutations (17, 19). Epigenetic inactivation by promoter methylation is increasingly being recognized as possibly the most common mechanism of transcriptional silencing. Biallelic methylation of the STK11 could certainly account for absent or markedly decreased labeling in these tumors. Additional postulated mechanisms of down-regulation include posttranscriptional instability of STK11 transcripts or post translational modification of the Stk11 products, with accelerated degradation. Interestingly, at least one of the cases with loss of Stk11 expression was an IPMN adenoma, suggesting that loss of expression may not be restricted to “late” lesions in IPMN progression (carcinoma in situ and invasive adenocarcinomas).
After establishing the specificity of the Stk11 antibody in terms of its ability to detect the STK11 gene product, we examined a large series of pancreatic (n = 56) and biliary (n = 38) cancers to elucidate the role of this tumor suppressor gene in the pathogenesis of these neoplasms. We found that a small proportion of pancreatic (7%) and biliary (2.6%) cancers demonstrates absent or markedly decreased Stk11 labeling; our initial assessments based on high-throughput screening of pancreatobiliary TMAs were subsequently validated on whole-tissue sections from the same cases. Given the markedly increased risk of familial pancreatic cancer in PJS patients harboring germline STK11 mutations, it is not surprising that a proportion of sporadic pancreatic adenocarcinomas also demonstrates abrogation of Stk11 function.
The relatively low frequency of Stk11 inactivation in pancreatic adenocarcinomas (4/56 or 7%) versus that reported in IPMNs (6/22 or 27%; P = .0519) suggests that there are genetic disparities between these two closely related neoplasms that may account for observed differences in clinical behavior. The genetic progression of IPMNs has not been completely defined. IPMNs are known to harbor mutations of p16 (20), p53(21) and DPC4(22), but the prevalence of the mutations is lower than that in ductal pancreatic adenocarcinoma. In fact, Dpc4 expression in IPMN is virtually normal unless associated with invasive ductal adenocarcinoma (23). STK11 is the first tumor-suppressor gene identified whose function may be more prevalently abrogated in IPMNs than in pancreatic ductal adenocarcinoma. The higher frequency of the STK11 inactivation observed in the IPMNs may be significant, and the possible contribution of the STK11 gene to the tumorigenesis of IPMNs would be worth further investigation.
CONCLUSION
In summary, we describe an antibody that we believe specifically detects the Stk11 protein and is a feasible surrogate for genetic analysis of STK11 status in archival sections. In addition, because immunohistochemistry can detect abnormalities irrespective of the mechanism of inactivation, this antibody has the potential to detect abnormalities of STK11 that are not demonstrable by mutational analysis alone. Future studies should focus on characterizing the expression of this protein in other sporadic cancers whose familial variants can occur with an increased frequency in PJS (e.g., pulmonary adenocarcinoma) and on determining the mechanisms of loss of Stk11 expression in the absence of coding-sequence mutations.
References
Jeghers HMV, Katz KH . Generalized intestinal polyposis and melanin spots of the oral mucosa, lip and digits: a syndrome of diagnostic significance. N Engl J Med 1949; 241: 1031–1036.
Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med 1987; 316: 1511–1514.
Hizawa K, Iida M, Matsumoto T, Kohrogi N, Kinoshita H, Yao T, et al. Cancer in Peutz-Jeghers syndrome. Cancer 1993; 72: 2777–2781.
Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, Burgart LJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med 1998; 128: 896–899.
Spigelman AD, Murday V, Phillips RK . Cancer and the Peutz-Jeghers syndrome. Gut 1989; 30: 1588–1590.
Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998; 391: 184–187.
Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 1998; 18: 38–43.
Su GH, Hruban RH, Bansal RK, Bova GS, Tang DJ, Shekher MC, et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999; 154: 1835–1840.
Sato N, Rosty C, Jansen M, Fukushima N, Ueki T, Yeo CJ, et al. STK11/LKB1 Peutz-Jeghers gene inactivation in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol 2001; 159: 2017–2022.
Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res 2002; 62: 3659–3662.
Guldberg P, thor Straten P, Ahrenkiel V, Seremet T, Kirkin AF, Zeuthen J . Somatic mutation of the Peutz-Jeghers syndrome gene, LKB1/STK11, in malignant melanoma. Oncogene 1999; 18: 1777–1780.
Wang ZJ, Churchman M, Campbell IG, Xu WH, Yan ZY, McCluggage WG, et al. Allele loss and mutation screen at the Peutz-Jeghers (LKB1) locus (19p13.3) in sporadic ovarian tumours. Br J Cancer 1999; 80: 70–72.
Karuman P, Gozani O, Odze RD, Zhou XC, Zhu H, Shaw R, et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell 2001; 7: 1307–1319.
Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP . Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1). Hum Mol Genet 2002; 11: 1497–1504.
Manley S, Mucci NR, De Marzo AM, Rubin MA . Relational database structure to manage high-density tissue microarray data and images for pathology studies focusing on clinical outcome: the prostate specialized program of research excellence model. Am J Pathol 2001; 159: 837–843.
Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992; 359: 235–237.
Abed AA, Gunther K, Kraus C, Hohenberger W, Ballhausen WG . Mutation screening at the RNA level of the STK11/LKB1 gene in Peutz-Jeghers syndrome reveals complex splicing abnormalities and a novel mRNA isoform (STK11 c.597(insertion mark)598 insIVS4). Hum Mutat 2001; 18: 397–410.
Resta N, Stella A, Susca FC, Di Giacomo M, Forleo G, Miccolis I, et al. Two novel mutations and a new STK11/LKB1 gene isoform in Peutz-Jeghers patients. Hum Mutat 2002; 20: 78–79.
Esteller M, Avizienyte E, Corn PG, Lothe RA, Baylin SB, Aaltonen LA, et al. Epigenetic inactivation of LKB1 in primary tumors associated with the Peutz-Jeghers syndrome. Oncogene 2000; 19: 164–168.
Moore PS, Orlandini S, Zamboni G, Capelli P, Rigaud G, Falconi M, et al. Pancreatic tumours: molecular pathways implicated in ductal cancer are involved in ampullary but not in exocrine nonductal or endocrine tumorigenesis. Br J Cancer 2001; 84: 253–262.
Sessa F, Solcia E, Capella C, Bonato M, Scarpa A, Zamboni G, et al. Intraductal papillary-mucinous tumours represent a distinct group of pancreatic neoplasms: an investigation of tumour cell differentiation and K-ras, p53 and c-erbB-2 abnormalities in 26 patients. Virchows Arch 1994; 425: 357–367.
Iacobuzio-Donahue CA, Klimstra DS, Adsay NV, Wilentz RE, Argani P, Sohn TA, et al. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: comparison with conventional ductal adenocarcinomas. Am J Pathol 2000; 157: 755–761.
Biankin AV, Biankin SA, Kench JG, Morey AL, Lee CS, Head DR, et al. Aberrant p16(INK4A) and DPC4/Smad4 expression in intraductal papillary mucinous tumours of the pancreas is associated with invasive ductal adenocarcinoma. Gut 2002; 50: 861–868.
Acknowledgements
Supported by The Michael Rolfe Foundation, The American Cancer Society (IRG 58–005–40), The Lustgarten Foundation Grant, the NIH (R03 CA93292), and the NIH (GI-SPORE CA62924).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sahin, F., Maitra, A., Argani, P. et al. Loss of Stk11/Lkb1 Expression in Pancreatic and Biliary Neoplasms. Mod Pathol 16, 686–691 (2003). https://doi.org/10.1097/01.MP.0000075645.97329.86
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000075645.97329.86
Keywords
This article is cited by
-
Regulation of metabolic reprogramming by tumor suppressor genes in pancreatic cancer
Experimental Hematology & Oncology (2020)
-
A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination
Laboratory Investigation (2019)
-
Regulation of triple-negative breast cancer cell metastasis by the tumor-suppressor liver kinase B1
Oncogenesis (2015)
-
Decreased LKB1 predicts poor prognosis in Pancreatic Ductal Adenocarcinoma
Scientific Reports (2015)
-
The genetic classification of pancreatic neoplasia
Journal of Gastroenterology (2015)
