
Abstract
The current classification system of renal tumors is based on morphologic criteria, as supported by genetic findings. We present a group of previously unclassified tumors with similar morphologic and genetic features, suggesting a new entity within renal neoplasms. Seven renal tumors from five patients (ages 31–67 years) were analyzed. All cases were stained with periodic acid–Schiff, Hale’s colloidal iron (HCI), and Alcian blue (AB) at pH 2.5/1.0 with and without hyaluronidase (HA) digestion. Immunohistochemical (IHC) stains were performed for CK8, CK18, CK19, vimentin, villin, Tamm-Horsfall protein (THP), renal cell carcinoma marker (RCC), epithelial membrane antigen (EMA), ulex europaeus agglutinin (UEA-1), soy bean agglutinin (SBA), peanut agglutinin (PNA), and MIB-1. Comparative genomic hybridization (CGH) and loss of heterozygosity (LOH) studies were performed on all cases. All tumors showed circumscribed growth, a tubular growth pattern with focal solid areas, no significant nuclear atypia and absence of necrosis, desmoplasia, or inflammation. Abundant extracellular mucin was present. Immunohistochemistry stains support collecting duct origin (EMA+, PNA+, SBA±, CK 8/18/19+, vimentin±, UEA-1−, RCC−, villin−, THP−). The proliferative rate was low (<1%). CGH showed multiple consistent chromosomal losses (−1,-4, −6, −8, −9, −13, −14, −15, −22). Clinical outcome was favorable, with recurrences but no known distant metastases or death of disease. These findings are distinct from all previously classified renal neoplasms. Our data suggest the presence of a unique tumor entity within tumors of probable collecting duct origin: tubular-mucinous renal tumors of low malignant potential.
Similar content being viewed by others
INTRODUCTION
Recent advances in pathology, especially those in the field of genetics and molecular diagnostics, have led to a new understanding of renal neoplasms. This is reflected in a new internationally accepted classification system, which is based on morphology and supported by common genetic findings (1, 2). However, about 6–7% of renal tumors remain currently unclassified.
Among renal epithelial tumors, collecting duct carcinomas (CDCs, Bellini duct carcinomas) are recognized as a rare but distinctive type of renal cell carcinoma (3, 4). They are generally considered to be aggressive tumors (4). Some reports, however, suggest a heterogeneous group of neoplasms, which is mainly based on morphologic, IHC, and clinical findings (3, 5). Consistent genetic alterations within this tumor group have not been well established (1).
We present a group of previously unclassified tumors with similar morphologic findings that suggest a new entity within renal neoplasms. Morphologic, immunohistochemical, and genetic findings strongly support that those tumors originate from collecting duct epithelium. Based on clinical findings, they are associated with a favorable prognosis.
MATERIALS AND METHODS
Seven renal tumors from five patients were evaluated. All cases were seen in consultation between 1994 and 2000. Clinical and pathologic findings were obtained from the referring pathologist and referring clinicians. In all cases, hematoxylin and eosin (HE)–stained glass slides with tumor and non-neoplastic renal tissue were examined. Paraffin-embedded tissue was available for ancillary studies in all cases.
Staining Procedures
Routine 4-μm sections from formalin-fixed, paraffin-embedded tissue were used for HE stains and histochemical and IHC evaluation. Histochemical stains were performed according to standardized protocols for periodic acid–Schiff, Hale’s colloidal iron, and Alcian blue (at pH 2.5 and pH 1.0). Alcian blue stains were performed with and without hyaluronidase digestion.
Immunohistochemistry
IHC stains were performed on paraffin-embedded tissues with antibodies to the following antigens: CK 8 (1:25), CK18 (1:25), CK19 (1:75), vimentin (1:25), MIB-1 (1:50), and EMA (1:50; all purchased from DAKO Diagnostika, Hamburg, Germany); RCC (1:50) and villin (1:100; both Novocastra, Newcastle upon Tyne, UK); UEA-1 (1:400, Vector, Burlingame); THP (1:50, Cedarlane, CA), SBA, and PNA (each 1:1000, Vector).
Briefly, after deparaffinization, tissue sections were microwaved twice for 7 minutes in 10 mwf citrate buffer (pH 6.0) and left in hot buffer at room temperature for an additional 30 minutes. For all stains, blocking of endogenous peroxidase was done with 3% H2O2 for 15 minutes. Sections were then placed in swine serum (diluted 1:10) for 10 minutes at room temperature. Primary antibodies were applied and allowed to react for 1 hour at room temperature. Sections were then incubated with a secondary biotinylated anti-mouse/anti-goat antibody and peroxidase-labeled streptavidin (LSAB2 System, DAKO). After color development with 3-amino-9-ethylcarbazole, the sections were counterstained with Hemalaun.
Mib-1 is a nuclear stain, and any staining of nuclei was considered positive. To determine a proliferative rate, 500 cells were counted in two randomly chosen high-power fields, and the percentage of positive nuclei was calculated. The remaining antibodies showed either cytoplasmic staining and/or membranous staining. Any focal staining equivalent to or stronger than an internal positive control was considered positive. Clearly visible staining of less than the positive control was considered as weak staining.
Comparative Genomic Hybridization
CGH was performed on all cases. For case 1, material for CGH was available from two tumors (1994, 2000). LOH studies were performed on three tumors (1978, 1994, 2000).
DNA Extraction
Paraffin-embedded tissue was cut into 10-μm sections, and areas of interest were microdissected with a scalpel blade. DNA extraction was performed with a commercially available kit (PeqLab, Erlangen, Germany).
Comparative Genomic Hybridization
FITC-labeled test DNA was hybridized to normal male metaphase chromosomes together with TRITC-labeled DNA from normal donors and unlabeled Cot-1 DNA, following a standard protocol (6). After counterstaining with DAPI, fluorescence images were captured, and the green:red ratios were calculated with a commercial software package (Applied Imaging).
Loss of Heterozygosity
Microsatellite markers (tetranucleotide repeats) were chosen from the Genome Database and amplified by PCR for 35 cycles. In each case, PCR products with tumor DNA and normal DNA were separated by polyacrylamide gel-electrophoresis and stained with ethidium bromide. For LOH studies, only those markers were chosen in which chromosomal localizations have already been confirmed by at least two independent research groups.
RESULTS
Clinical Data
All patients in our series were female. The age range at the time of tumor resection was 31–67 years. In four cases, a nephrectomy was performed. Three tumors were resected by partial nephrectomy. Six of seven tumors were organ confined (pT1 or pT2). Follow-up information was available for three patients. Two patients were alive, with no evidence of disease (NED), 16 and 33 months after resection. One patient (Case 1) has been followed since 1978. Since then, she has had three tumors with similar morphology removed from the right and left kidney. Likewise, she is currently alive with NED, 23 years after an initial tumor resection (Table 1).
Gross Findings
Tumor size varied from 2.8 to 10 cm. All tumors were sharply circumscribed, with a homogenous yellow to tan brown to pinkish cut surface. In two cases, focal hemorrhage was seen. There was no evidence of renal vein invasion. In two cases, a compression of the renal pelvis without tumor infiltration was noted.
Microscopic Findings
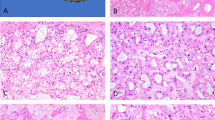
Histology showed circumscribed growth in all cases. Tumors were partly surrounded by a small rim of compressed fibrous tissue with scattered lymphocytes (Fig. 1). There was no intratumoral lymphocytic or neutrophilic inflammation. All tumors consisted of epithelial cells with few intervening stroma. The predominant architectural pattern was that of vague tubular growth (Fig. 2, left). Tubules were lined by cuboidal cells with pale to eosinophilic cytoplasm. Tumor cells contained centrally located round nuclei without significant nuclear atypia. Most nuclei contained easily discernible small to medium-sized nucleoli (Fig. 2, right). All tumors displayed focal areas of solid growth. This was frequently associated with tumor cells of a more spindled appearance, although cellular features differed from those of a sarcomatoid growth pattern (Fig. 3). A striking feature in all cases was the presence of abundant, predominantly extracellular mucin (Fig. 4, left). Occasional mucin lakes were noted to expand stroma between tumor cells. Also, all cases included small clusters of foamy macrophages that were usually associated with mucin collections (Fig. 4, right). Mucin collections and macrophages were seen in association with both tubular and solid growth patterns. Two tumors contained areas of focal hemorrhage. One tumor showed focal perirenal fat invasion (Case 3). None of the tumors displayed areas of necrosis or desmoplasia with neutrophilic inflammation. Also, none of the cases had a cystic or a papillary growth pattern with fibrovascular cores or psammoma bodies.

Circumscription.

Tubular growth (left); nuclear features (right).

Spindled growth pattern.

Extracellular mucin (left); collections of macrophages (right).
Histochemical Stains
Abundant extracellular mucin of acid epithelial type was present in all cases. Mucin was strongly positive for Alcian blue at pH 2.5. All cases showed moderate or strong staining at pH 1.0 with and without hyaluronidase digestion, as well as with Hale’s colloidal iron stain (Fig. 5). Extracellular mucin was periodic acid-Schiff negative. Rare periodic acid-Schiff–positive intracellular droplets were noted.

Histochemical stains.
Immunohistochemical Stains
The proliferative rate (MIB-1) was low (<1%), with one to five positive nuclei within 500 cells counted. In four cases, positive nuclei were randomly distributed within the tumor. In one case, positive nuclei were more frequently seen peripherally.
All tumors were positive for CK 8/18. Four of five tumors were weakly positive for CK19, two of those cases showed additional strong staining of rare peripherally located neoplastic tubules. Two of five cases were focally positive for vimentin.
IHC stains for EMA and PNA were positive in all five cases, with at least focally strong staining in tubular areas (Fig. 6). Staining was predominantly apical and membranous. Focal weak cytoplasmic staining was also noted. Areas with solid growth patterns within the same tumor showed decreased or absent staining. SBA was focally positive in three of five cases, with an apical and cytoplasmic staining pattern. Also, those three cases showed occasional staining of small cytoplasmic vacuoles. UEA-1 was consistently negative.

Immunohistochemical stains.
All tumors were negative for RCC and villin, two markers of proximal tubules. Also, all tumors were negative for THP, a marker of the thick ascending loop of Henle and distal convoluted tubules. Results are summarized in Table 2.
Comparative Genomic Hybridization
All tumors had multiple genetic alterations that included consistent losses of chromosomes 1, 4, 6, 8, 9, 13, 14, 15, and 22 (Fig. 7). These findings were confirmed by LOH studies in all cases that had polymorphisms within normal renal epithelium. For Case 1, CGH was performed on two tumors, resected in 1994 and 2000. Both tumors had identical chromosomal alterations. Material from a tumor resected in 1978 was available, but quality was inadequate to perform CGH. For this tumor, only LOH studies were performed. LOH results indicate that all three tumors originated from the same clone (Fig. 8). CGH results are summarized in Table 3.

Case 1, CGH (tumor resected in 2000). Red color indicates loss of genomic material (male reference).

Case 1, LOH studies of chromosome 1.
DISCUSSION
We present seven renal epithelial tumors with unusual morphologic features. All cases were diagnostically challenging and were seen in consultation by one of the authors (SS). Within the currently used classification system of renal tumors, those cases are considered to be unclassified renal neoplasms (2). Lack of tumor necrosis, a low proliferative activity, and nuclear features suggest tumors of low malignant potential. Immunohistochemical and genetic findings strongly support origin from collecting duct epithelium.
Histologically, all tumors had tubular and solid growth patterns with focally extensive extracellular mucin production, as described in some “classical” CDCs (4). However, there was a lack of other characteristic morphologic features seen in classical CDCs, such as infiltrative growth, pleomorphic cells with significant nuclear atypia, a cystic component with “hobnail” epithelium, desmoplasia, or intratumoral neutrophilic inflammation (3, 4). Immunohistochemical staining patterns were similar in all cases: no staining was noted with brush-border–associated antigens of proximal tubules, such as villin or RCC (7, 8). Also, no staining was seen with THP, a marker that is found in distal convoluted tubules and the ascending loop of Henle (7). Positive staining was seen with markers that are found in collecting duct epithelium (3, 7, 9), including EMA and PNA. Three tumors showed additional focal staining with SBA. UEA-1, a marker that has been reported to be positive in some but not all CDCs, was negative in all cases (3, 4, 5).
Genetic alterations were consistent in all cases. They included losses of chromosomes 1, 4, 6, 8, 9, 13, 14, 15, and 22. Recently, it has been recognized that distinctive types of renal tumors also have common documented genetic abnormalities (1). Those correlate with histologic findings and support the currently used classification system of renal cell tumors (2). Tumors that originate from collecting duct epithelium, including chromophobe RCC and oncocytomas, show frequent chromosomal losses (10). In chromophobe RCC, monosomies of chromosomes 1, 2, 6, 10, 13, 17, and 21 have been described by several authors (11, 12). In oncocytomas, losses of chromosome 1 and Y have been reported (13, 14). Both tumors originate from intercalated cells (cortical collecting duct epithelium). CDCs, or Bellini duct carcinomas, arise from medullary collecting duct epithelium (3). They comprise <1% of all renal epithelial tumors, and therefore cytogenetic data are scarce. To date, no consistent pattern of genetic abnormalities has been established. Fuezesi et al. (15) was one of the first investigative groups to karyotype CDCs. They found losses of chromosomes 1, 6, 14, 15, and 22. These genetic alterations are similar to our own findings. Based on those investigators’ published photos (of tubulopapillary tumors with psammoma bodies), however, results were found in tumors that do not match the morphology of classical CDC. Therefore, we doubt that those investigators analyzed cases of classical CDC. Although several other authors have also investigated CDCs, none of them could reproduce the findings of Fuezesi et al. (15) Their results were less consistent and included variable chromosomal gains and losses (16, 17). Most consistent alterations include losses of 8p and 13q (1, 18). Furthermore, we have performed CGH on two “typical” CDCs from our own files. Likewise, this has returned different genetic alterations (data not shown).
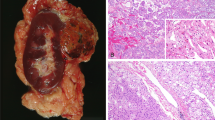
CDC are generally considered to be aggressive neoplasms, with early recurrence, metastasis and frequent death (3). Although our IHC findings suggest a tumor of collecting duct origin, morphologic features favor a low-grade malignant phenotype. Accordingly, we have not seen aggressive behavior in CDCs in our patients. Follow-up data were available in three cases. Two patients were alive with NED, 16 and 33 months after initial diagnosis. One patient (our Case 1) was followed clinically over 23 years. She was initially diagnosed with a “renal adenoma” that was resected in 1978. She presented with a second tumor in 1994 and underwent partial nephrectomy for a third renal tumor of the contralateral kidney in 2000. All three tumors had very similar morphologic features (Fig. 9). Currently this patient is alive, with no evidence of disease. CGH revealed identical genetic alterations in those tumors that were resected in 1994 and 2000. Furthermore, LOH studies were performed on all three tumors. Results indicate that all three tumors originated from the same clone.

Case 1, histology of three tumors resected between 1978 and 2000.
It has already been established that a spectrum of low-grade and high-grade neoplasms does exist within tumors of collecting duct origin. Chromophobe RCCs are considered to be nonaggressive (cortical) collecting duct neoplasms. Could our presented tumors be variants of chromophobe RCCs or even oncocytomas? Based on morphologic findings alone, a relation between those tumors seems unlikely. There is some overlap of genetic alterations. Overall, however, both tumors have different genetic profiles. Chromophobe RCCs include losses of chromosomes 2, 10, and 17. Also, tetraploidization may occur (11). Both features were not found in any of our tumors.
Only few data on low-grade variants of medullary CDC are published. MacLennan et al. (5) have described 13 cases of low-grade mucinous tumors of possible collecting duct origin. Those investigators describe cases with solid and cystic architecture. In our own series, all tumors show tubular and solid-growth patterns without cyst formation. Srigley et al. (19) presented a poster abstract of seven tumors with similar morphology, suggesting origin from the loop of Henle. Another poster abstract with four published cases of similar morphology suggests distal nephron differentiation (20). To our knowledge, genetic data are not available in any of those cases; nevertheless, we think that at least some of those may represent neoplasms that belong to our own described tumor entity.
Could our tumors represent benign tumors, that is, adenomas? We find this unlikely, although we cannot entirely exclude this possibility. There are some clinical and morphologic similarities with metanephric adenomas, which are frequently reported in women (21, 22). Interestingly, tumors in our own study occurred exclusively in female patients. Based on our small numbers of five patients, however, we cannot be sure whether this may represent an incidental finding. Morphologically, metanephric adenomas may display areas with tubular or acinar growth patterns. Frequently though, there are additional histologic findings, such as papillary growth or psammoma bodies. About 50% of cases also contain glomeruloid bodies. None of these features was present in our tumors. Also, most authors report an absence of immunohistochemical staining for EMA (21, 22, 23) which was positive in all of our tumors. Few groups have investigated metanephric adenomas for genetic alterations. Although most authors found a normal diploid karyotype (23, 24), one study has shown trisomies 7 and 17 (25). Recently, a tumor suppressor gene has been mapped to chromosome 2p13 (26). Overall, there are no reported clinical recurrences for metanephric adenomas (22).
Based on histology, renal tubulopapillary adenomas may also enter the differential. These tumors may rarely present with an entirely tubular growth pattern. Again though, most cases display at least focal papilllary growth patterns and frequently contain psammoma bodies. Although one study showed frequent positive IHC staining for EMA and PNA (27), most expressed antigens are those found in proximal tubules (28). More important, tubulopapillary adenomas are small lesions that rarely exceed 5 mm. Cytogenetic alterations appear to be limited to +7, +17, and -Y (29).
Conversely, all tumors in our own study had major genetic alterations. This is unusual for entirely benign lesions. We have not seen any distant metastasis, but one patient had three intrarenal tumors with identical histologic and immunohistochemical features removed within 23 years, all originating from the same clone. We therefore conclude that these neoplasms are best assigned as tumors of low-malignant potential, until further information is available about their long-term behavior.
In summary, we present a group of uncommon renal tumors (Table 4). All patients were female. The tumors were all solid and well circumscribed on gross examination. Histology showed a predominantly tubular growth pattern with low- to intermediate-grade nuclear morphology and abundant extracellular mucin production. All cases displayed a similar immunohistochemical staining pattern, supporting collecting duct origin (EMA+, PNA+). Genetic alterations within these tumors were consistent and also different from all other defined renal tumor entities, including those of collecting duct origin. Follow-up data with no known distant metastases support a favorable prognosis. Based on these findings, we suggest a new entity of renal tumors of collecting duct origin: tubular-mucinous renal tumors of low malignant potential.
References
Kovacs G, Akhtar M, Beckwith BJ, et al. The Heidelberg classification of renal cell tumours. J Pathol 1997; 183: 131–133.
Stoerkel S, Eble JN, Adlakha K, et al. Classification of renal cell carcinoma: workgroup no. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer 1997; 80: 987–989.
Amin MB, Varma MD, Tickoo SK, et al. Collecting duct carcinoma of the kidney. Adv Anat Pathol 1997; 4: 84–95.
Srigley JR, Eble JN . Collecting duct carcinoma of the kidney. Semin Diagn Pathol 1998; 15: 54–67.
MacLennan GT, Farrow GM, Bostwick DG . Low-grade collecting duct carcinoma of the kidney: report of 13 cases of low-grade mucinous tubulocystic renal carcinoma of possible collecting duct origin. Urology 1997; 50: 679–684.
Stoerkel S, Simon R, Brinkschmidt C, et al. Komparative genomische Hybridisierung in der Pathologie. Pathologe 1996; 17: 189–194.
Moll R . Vorbemerkungen zur Morphologie des Nephrons. In: Moll R, editor. Cytokeratine als Differenzierungsmarker: expressionsprofile von Epithelien und epithelialen Tumoren. Stuttgart, Germany: Gustav Fischer Verlag.
Yoshida SO, Imam A . Monoclonal antibody to a proximal nephrogenic renal antigen: immunohistochemical analysis of formalin-fixed paraffin-embedded human renal cell carcinomas. Cancer Res 1989; 49: 1802–1809.
Ulrich W, Horvat R, Krisch K . Lectin histochemistry of kidney tumours and its pathomorphological relevance. Histopathology 1985; 9: 1037–1050.
Reuter VE, Presti JC Jr . Contemporary approach to the classification of renal epithelial tumors. Semin Oncol 2000; 27: 124–137.
Gunawan B, Bergmann F, Braun S, et al. Polyploidization and losses of chromosomes 1, 2, 6, 10, 13, and 17 in three cases of chromophobe renal cell carcinomas. Cancer Genet Cytogenet 1999; 110: 57–61.
Speicher MR, Schoell B, du Manoir S, et al. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am J Pathol 1994; 145: 356–364.
Brown JA, Takahashi S, Alcaraz A, et al. Fluorescence in situ hybridization analysis of renal oncocytoma reveals frequent loss of chromosomes Y and 1. J Urol 1996; 156: 31–35.
Steiner G, Sidransky D . Molecular differential diagnosis of renal carcinoma. Am J Pathol 1996; 149: 1791–1795.
Fuezesi L, Cober M, Mittermayer C . Collecting duct carcinoma: cytogenetic characterization. Histopathology 1992; 21: 155–160.
Gregori-Romero MA, Morell-Quadreny L, Llombart-Bosch A . Cytogenetic analysis of three primary bellini duct carcinomas. Genes Chromosomes Cancer 1996; 15: 170–172.
Verdorfer I, Culig Z, Hobisch A, et al. Characterization of a collecting duct carcinoma by cytogenetic analysis and comparative genomic hybridization. Int J Oncol 1998; 13: 461–464.
Schoenberg M, Cairns P, Brooks JD, et al. Frequent loss of chromosome arms 8p and 13q in collecting duct carcinoma (CDC) of the kidney. Genes Chromosomes Cancer 1995; 12: 76–80.
Srigley JR, Eble JN, Grignon DJ, et al. Unusual renal cell carcinoma (RCC) with prominent spindle cell change possibly related to the loop of Henle [abstract]. Mod Pathol 1999; 12: 107A.
Parwani AV, Husain AN, Epstein JI, Beckwith JB, Argani P . Low grade mucinous renal carcinomas with distal nephron differentiation [abstract]. Mod Pathol 2001; 14: 120A.
Davis CJ, Barton JH, Sesterhenn IA, et al. Metanephric adenoma. Clinicopathological study of fifty patients. Am J Surg Pathol 1995; 19: 1101–1114.
Grignon DJ, Eble JN . Papillary and metanephric adenomas of the kidney. Semin Diagn Pathol 1998; 15: 41–53.
Gatalica Z, Grujic S, Kovatch A, et al. Metanephric adenoma: histology, immunophenotype, cytogenetics, ultrastructure. Mod Pathol 1996; 9: 329–333.
Granter SR, Fletcher JA, Renshaw AA . Cytologic and cytogenetic analysis of metanephric adenoma of the kidney: a report of two cases. Am J Clin Pathol 1997; 108: 544–549.
Brown JA, Anderl Kl, Borell TJ, Qian J, Bostwick DG, Jenkins RB . Simultaneous chromosome 7 and 17 gain and sex chromosome loss provide evidence that renal metanephric adenoma is related to papillary renal cell carcinoma. J Urol 1997; 158: 370–374.
Pesti T, Sukosd F, Jones EC, Kovacs G . Mapping a tumor suppressor gene to chromosome 2p13 in metanephric adenomas. Hum Pathol 2001; 32: 101–104.
Hiasa Y, Kitamura M, Nakaoka S, et al. Antigen immunohistochemistry of renal cell adenomas in autopsy cases: relevance to histogenesis. Oncology 1995; 52: 97–105.
Stoerkel S . Karzinome und Onkozytome der Niere. Stuttgart, Germany: Gustav Fischer Verlag, 1993.
Kovacs G . The value of molecular genetic analysis in the diagnosis and prognosis of renal cell tumours. World J Urol 1994; 12: 64–68.
Acknowledgements
The authors thank Ms. Böhmer and Ms. Sauer for their expertise with the applied technical procedures. Also, they thank the following referrers and institutions for their case submission and support with follow-up data: Prof. Dr. Mihatsch, Universität Basel (Switzerland); Prof. Schmid, Universität Essen (Germany); PD Dr. Theissig, Technische Universität Dresden (Germany); and Dr. Fries, Universität Köln (Germany).
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented in part at the 90th Annual Meeting of the United States and Canadian Academy of Pathology in Atlanta, GA, March 2001.
Rights and permissions
About this article
Cite this article
Rakozy, C., Schmahl, G., Bogner, S. et al. Low-Grade Tubular-Mucinous Renal Neoplasms: Morphologic, Immunohistochemical, and Genetic Features. Mod Pathol 15, 1162–1171 (2002). https://doi.org/10.1097/01.MP.0000031709.40712.46
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000031709.40712.46
Keywords
This article is cited by
-
Mucinous tubular and spindle cell carcinoma of the kidney: a report of seven cases
BMC Cancer (2023)
-
Mucinous tubular and spindle cell carcinoma with high-grade transformation: case report
Surgical and Experimental Pathology (2022)
-
Adverse histology, homozygous loss of CDKN2A/B, and complex genomic alterations in locally advanced/metastatic renal mucinous tubular and spindle cell carcinoma
Modern Pathology (2021)
-
The spectrum of renal cell carcinoma in adults
Abdominal Radiology (2016)
-
Mucinous tubular and spindle cell renal cell carcinoma: a review of clinicopathologic aspects
Diagnostic Pathology (2015)
