
Abstract
The cytogenetic abnormalities of granulosa cell tumors (GCT) of the ovary are only partially known. Up to now, mainly numerical chromosomal aberrations have been described. Therefore we performed a comprehensive study on paraffin-embedded material of 20 GCT (17 adult, 3 juvenile; patient age between 16 and 78 y) combining comparative genomic hybridization (CGH); fluorescence in situ hybridization (FISH) using DNA-specific probes for chromosome 12, 17, 22, and X; DNA cytometry; and immunohistochemistry (inhibin, p53, Ki67).
By DNA cytometry, 16 of 20 tumors (80%) were diploid. However, 6 of 16 diploid tumors (37%) showed aberrations by FISH. FISH revealed monosomy 22 in 8/18 cases (40%); trisomy 12 in 5/20 (25%); monosomy X in 2/20 (10%); and loss of chromosome 17 in one case (5%). The main findings by CGH were gains of chromosomes 12 (6 cases, 33%) and 14 (6 cases, 33%) and losses of chromosomes 22 (7 cases, 35%) and X (1 case, 5%), mostly comprising whole chromosomes or chromosome arms. Inhibin and p53 were expressed in 100% and 95% of the tumors, respectively. The Ki67 index ranged from 0% to 61%. Neither immunohistochemistry, nor DNA cytometry and molecular genetic analysis, provided statistically significant prognostic information.
In summary, our study reveals a distinctive pattern of cytogenetic alterations in GCT. Our observations confirm earlier reports that trisomy 12 and 14 are frequent aberrations; however, monosomy 22 seemingly is even more prevalent.
Similar content being viewed by others
INTRODUCTION
Granulosa cell tumors of the ovary (GCT) are much less common than epithelial ovarian cancer. These tumors account for 1.5% of all ovarian neoplasms and for 6% of malignant ovarian tumors. Sixty-five percent of patients are postmenopausal; <5% are prepubertal. The juvenile granulosa cell tumor is a distinctive form of GCT that occurs almost exclusively in children and young adults. More than 95% of the GCT are unilateral and confined to the ovary.
GCT of the ovary is considered a low-grade malignancy. In most cases, the clinical course is nonaggressive and the 10-year survival rate is >90%. Tumors that exhibit mitotic figures or cellular atypia may behave more aggressively. Available information about the prognostic value of these factors, as well as age, histological type, and clinical stage is mostly incomplete and controversial (1, 2, 3, 4, 5, 6).
The DNA index of GCT is mostly in the diploid range (7). Only limited numbers of GCT tumors (adult and juvenile type) have been analyzed by traditional cytogenetics. Recent examinations of archival material by FISH have failed to confirm the high prevalence of trisomy 12 initially thought to be present in GCT and other sex cord–stromal tumors (8, 9, 10). Several other aberrations including monosomy 22, trisomy 14, and rearrangement of 6q have been reported (11). To the best of our knowledge, studies using comparative genomic hybridization (CGH) have not been reported so far for GCT.
Immunohistochemistry can be useful in the differential diagnosis of GCT. Inhibin is probably the most helpful marker (2, 12, 13, 14, 15, 16, 17, 18, 19). Inhibin is a dimeric glycoprotein hormone produced by normal ovarian granulosa cells and testicular Sertoli cells (20) that is involved in the regulation of pituitary follicle-stimulating hormone secretion. Elevated levels of inhibin have been found in the serum of patients with granulosa cell tumors (21, 22). Whether immunohistochemical analysis of the proliferation-associated antigen Ki67 and the tumor suppressor p53 gene plays a prognostic role in GCT is not fully clarified (23).
In the present study we therefore performed a comprehensive analysis of 20 GCT combining fluorescence in situ hybridization (FISH), CGH, DNA cytometry, and immunohistochemistry. The results were compared with the clinical outcome of the patients. The study aimed at a better characterization of genetic alterations in these tumors that might be relevant for diagnosis, prognosis, and treatment decisions.
MATERIALS AND METHODS
Archival material of 20 granulosa cell tumors was studied, including three of juvenile type (Cases 13, 19, 20) and 17 of adult type. The age ranged from 16 to 78 years (mean age, 53.4 y). All cases were staged according to the criteria of the International Federation of Gynaecologists and Obstetricians (FIGO) and the International Union against Cancer (UICC). FIGO Ia was represented in 11 cases (55%); FIGO Ic, in 4 cases (20%); and FIGO IIIc, in 5 cases (25%). Per case, between 1 and 15 representative histological slides were available.
All patients were treated surgically between 1985 and 2000 at the same institution (Department of Gynaecology of the Klinikum Grosshadern, University of Munich). No patient received any other treatment before operation. Fifteen of 20 cases had complete clinical and pathologic information, including follow-up data. The average length of follow-up was 7.2 years.
Comparative Genomic Hybridization
Cell culture, chromosome preparation and karyotype analysis were performed as previously described (24). One microgram of tumor DNA was labeled by a standard nick-translation reaction with biotin-16-dUTP and 700 ng reference DNA from a healthy male donor with digoxigenin-11-dUTP (each from Boehringer Mannheim). The purification of labeled DNA fragments of <100 base pairs was achieved by applying column chromatography (Sephadex-G50). Repetitive sequences were blocked with 70 μg of Cot 1 DNA.
Gray-band images of DAPI, FITC, and rhodamine fluorescence were taken using a CCD camera coupled to a Zeiss microscope. For digital image analysis and subsequent karyotyping, the QUIPS-XL (Applied Imaging, Vysis, Downers Grove, IL) software package was used. Only metaphase spreads showing a high green-to-red intensity were taken in account. Between 6 and 18 metaphases (mean = 10.2) for each chromosome were analyzed. Corresponding ratio profiles were evaluated only within the 99% confidence limit. The 50% thresholds (upper threshold, 1.2; lower threshold, 0.8) were applied to define the chromosomal regions of DNA sequence losses and gains.
Fluorescence In Situ Hybridization
Two selected tissue blocks for each case were used. Under microscopic control, excess normal tissue was removed by microdissection, and tumor cell nuclei were extracted by the method of Hyytinen et al. (25). The in situ hybridization was performed as described previously (26). The hybridization mix was prepared according to the manufacturer's protocol with DNA-specific probes for chromosomes 12, 17, 22, and X (Vysis, Downers Grove, IL).
The slides were evaluated under a Zeiss (Oberkochen, Germany) Axioscop fluorescence microscope equipped with an HBO-100W mercury lamp and dual and triple band-pass filters (Vysis). Hybridization signals were counted in 200 nuclei per case. After exclusion of all overlapping nuclei, only discrete signals in nuclei with a distinct nuclear border were evaluated.
Image Cytometry
Image cytometry (ICM) was performed on Feulgen-stained pellets of the nuclear suspensions prepared for FISH analysis in all 20 cases (total of 40 pellets, on average 2 per case). For the measurements, a PC-based image analyzer, ACAS (Ahrens, Bargteheide, Germany) with special DNA software was used. For assessment of the diploid (2c) range, the integrated optical density of the nuclei of 25–30 granulocytes in the pellets was analyzed. The CV value of these reference cells was not to exceed 6%. Subsequently, an average of 200 (185–217) tumor cell nuclei was measured selectively in each sample. Only intact, nonoverlapping nuclei were included in the analysis.
Aneuploidy was assumed when >45% of the nuclei exhibited a DNA content above 2.5c or when >15% showed DNA values higher than 5c (2.5c- and 5c-exceeding rate).
Immunohistochemistry
All paraffin-embedded specimens were cut at 2–3 μm and mounted on SuperFrost/Plus microscope slides (Menzel, Germany). After deparaffinization and rehydration immunohistochemical assays were performed by standard methods.
For inhibin mouse anti-human inhibin 32-kDa alpha subunit, clone R1 (Serotec, Kidlington, Oxford, UK) was used at a dilution of 1: 40. The binding of primary antibodies was detected by use of the alkaline phosphatase-anti-alkaline phosphatase (APAAP) method. Mayer's hematoxylin was used as counterstain. A brown reaction product in the cytoplasm indicated specific staining. As positive control, tissue sections of a corpus luteum were used.
The immunohistochemical results of inhibin were scored semiquantitatively, resulting in four groups: negative (0 positive cells), weak (1–10% positive cells), moderate (11–50% positive cells), and strong staining (51–100% positive cells).
For Ki67, the monoclonal antibody MIB1 (Dakopatts, Glostrup, Denmark; DAKO) was used at a dilution of 1:50. The binding of primary antibodies was detected by use of the alkaline phosphatase–anti-alkaline phophatase (APAAP) method (DAKO). Fast red was used as counterstain.
The immunohistochemical results of Ki67 were scored quantitatively. One thousand cells in each slide were counted. The percentage of the immunoreactive tumor cells was determined and expressed in 5% increments.
The monoclonal antibody against p53 (DAKO) was used at a dilution of 1:100. The binding of primary antibodies was detected by use of the alkaline phosphatase–conjugated streptavidin (super-sensitive label) method (DAKO). Negative controls were performed by replacing the primary monoclonal antibodies with RPMI.
p53 accumulation was assumed if >10% of the tumor cells showed a nuclear immunoreaction and then was scored in three groups, according to the protocol of Gebhart et al. (23): weak (<20%), moderate (20–60%), and strong (>60%).
Statistic Analysis
For all statistical calculations, SPSS 10 statistical software (Superior Performance Software System, SPSS for Windows version 7.0, Microsoft, Redmond, WA) was used. The various parameters were compared by χ2 tests; Kaplan-Meier-survival curves were analyzed by log rank test. P < .05 was regarded as statistically significant.
RESULTS
Image Cytometry
In all cases under study (n = 20), the determination of DNA ploidy was possible by means of ICM (mean CV values were 3.5% and 3% for all diploid and nondiploid peaks, respectively). In the ICM analysis, 16 of 20 cases (80%) were diploid, and 4 (20%: 3 adult, 1 juvenile), nondiploid (15% tetraploid and 5% aneuploid). There was no correlation of DNA ploidy with patients' age and tumor stage.
Fluorescence In Situ Hybridization
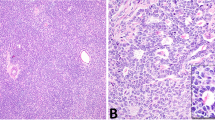
FISH analysis of nuclear suspension was successful in all cases for chromosome 12, 17, and X and in 18 cases (90%) for chromosome 22. Eight of 18 cases (44%) showed monosomy 22; five of 20 cases (25%), trisomy 12; 2 of 20 cases (10%), monosomy X; and one case (5%), monosomy 17. Two cases of monosomy 22, three cases of trisomy 12, and one case of monosomy X were DNA-aneuploid. Of the 16 DNA-diploid GCT, 6 cases (34%) showed aberrations by FISH. Two examples are represented in Figure 1.

Fluorescence in situ hybridization analyses. A, example for trisomy 12. (Four nuclei display three hybridization signals.) In one nucleus, two signals are split spots. B, example for monosomy 17. Two nuclei contain one and two hybridization signals, respectively.
Comparative Genomic Hybridization
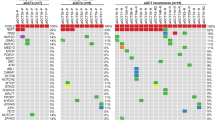
CGH analysis was possible in all cases. The results of FISH and CGH with regard to chromosomal losses and gains are summarized in Figure 2. Genetic changes were seen in 75% (n = 15). Almost all chromosomes were affected. The most frequently occurring CGH changes were complete loss of chromosomes 22 (40%), 21 (10%), and 4 (10%). Complete loss of chromosomes 5, 13, 16, 17, 18, 19, and X was seen once each (5%). Complete gains of chromosomes were noticed for chromosome 14 (33%), 12 (15%), 8 (10%), 9 (10%), 15 (10%), and 21 (10%) and in one case each (5%) for chromosomes 3, 7, 10, 18, and 20. Partial losses and gains were seen on nearly every chromosome (see Table 1 and Fig. 2).

Ideogram showing comparative genomic hybridization findings in 20 granulosa cell tumors (red bars on the left of the chromosomes are losses; green bars on the right are gains).
Comparison of FISH and CGH results showed that 7 of 8 (87%) losses of chromosome 22 seen by FISH could be confirmed by CGH. Three of 5 cases (60%) with trisomy 12 in FISH analysis revealed gains of the whole chromosome 12. The other two cases and one additional case showed partial gains on chromosome 12 in CGH analysis. In contrast, FISH monosomy 17, seen in a single case, was not confirmed by CGH, and only one of two FISH monosomies X (50%) was also observed by CGH. CGH did not detect any additional changes on these four chromosomes, which were not revealed by FISH, with the exception of Case 10, which showed a loss of centromeric signals of chromosomes 17 and 22 by FISH that could not be detected by CGH.
Immunohistochemistry
Inhibin expression was found in all cases. It was moderate or strong in 16 (80%) and weak in 4 tumors (20%). An example is given in Figure 3A.

Immunohistochemical detection of moderate inhibin expression (A), nuclear p53 accumulation (B), and Ki67 (C). Magnification each: 40 × objective.
Nuclear expression of the proliferation-associated antigen Ki67 was detected in 18 tumors (90%). In half of the cases (n = 10), the Ki67 index was <5%; in 9 tumors (45%), between 5 and 25% of the tumor cell nuclei were stained. One case of a juvenile granulosa cell tumor showed a proliferation fraction of 61% (Fig. 3B).
Nuclear p53 accumulation was seen in 18 of 19 tumors (94.7%). One case could not be evaluated; one did not show any expression. Three cases showed p53 in <20% nuclei; 9 cases, in 20–60% of the tumor cell nuclei; and 6 cases expressed p53 in >60% of tumor cells (Fig. 3C). p53 expression and Ki67 index did not correlate.
Juvenile GCT
Two of three juvenile GCT (66%) showed low proliferation indices (Ki67) of 8 and 17% and moderate p53 accumulation (60% each). Being diploid in DNA cytometry, these two cases did not show any changes by FISH and CGH analysis. The third juvenile GCT (Case 19) displayed a high Ki67 index (61%) and strong p53 expression. It was DNA aneuploid and showed a large number of chromosomal gains and losses (Table 1).
Survival Analysis
For 15 cases (13 adult and two juvenile), complete follow-up data were available. Four adult patients died of disease. p53, Ki67, and DNA ploidy were not correlated with outcome. Furthermore, neither the number of cytogenetic aberrations nor specific chromosomal changes had prognostic significance for both tumor types, the adult and juvenile GCT.
DISCUSSION
Granulosa cell tumor of the ovary is an uncommon and sometimes misdiagnosed tumor (27) of low malignant potential. Therefore, accurate diagnosis of these tumors is crucial for optimal treatment decisions, which mainly depend on tumor stage and age of the patient. Tumor stage is the only established prognostic factor. The prognosis for Stage IA patients is excellent, and fortunately most cases are Stage IA. On the other hand, long-term survival is poor in patients with extraovarian spread. The biological factors that determine tumor behavior and prognosis are largely unknown. Therefore, in the present investigation we examined 20 cases of adult and juvenile GCT combining molecular pathology, DNA cytometry, and immunohistochemistry.
Image cytometry revealed DNA diploidy in 80%, which is in agreement with observations of Halperin et al. (28), Gorski et al. (29), and Suh et al. (30). Several groups of investigators have reported that diploidy is associated with favorable prognosis (31, 32, 33, 34, 35); however, in the present study ploidy and survival were not correlated. It has been pointed out (27, 36) that image cytometric analysis is not sensitive enough to detect small changes in nuclear DNA content. Our results support this notion. Our combined FISH and CGH study revealed chromosomal aberrations in 11 of 16 DNA diploid cases (68%).
There are no universally accepted cutoffs for FISH diagnosis of chromosomal and/or gene gains or losses. Although our definitions of gene aberrations may be viewed as more or less subjective, the high concordance of FISH and CGH results is reassuring and suggests that the cutoffs used in our study are reliable.
Gains of whole chromosomes were the most characteristic chromosomal aberration observed in the present series of GCT. A number of previous studies noted trisomy 12 to be a frequent abnormality in tumors of the female genitourinary tract. It is a relatively common finding in sex cord–stromal tumors (11, 28, 29, 37, 38, 39, 40). In agreement with these studies, we detected trisomy 12 by FISH analysis in five GCT (25%). By CGH, six cases showed gains of the whole chromosome 12 or at least of the short arm. In contrast to the results of Halperin et al. (28), we did not see an association of trisomy 12 with the juvenile type of GCT. Several important genes have been identified on chromosome 12 (e.g., KRAS 2, KRAG, MDM2); however, it is not clear whether they play a role in GCT.
In the present series, monosomy 22 was more prevalent than trisomy 12. It was found in 8 of 20 GCT (40%), suggesting that it represents a nonrandom change (39, 40, 41). Like Van den Berghe et al. (41), we observed that monosomy was frequently associated with other chromosomal abnormalities, mostly with trisomy 14 (5/8 cases, 62%). Furthermore, two of four patients who died of disease had monosomy 22 (50%). Thus, loss of chromosome 22 may be associated with tumor progression of GCT.
NF2 is a well-characterized tumor suppressor gene on chromosome 22; however, mutations of NF2 in GCT have not been reported so far. In other tumors like breast, liver, and colorectal carcinomas, NF2 does not appear to play a major role. Therefore, the existence of other tumor suppressor loci has been proposed. These postulated tumor suppressor genes may also be involved in GCT (39).
The first report of trisomy 14 in GCT was by Gorski et al. (29). In our study, we saw gain of chromosome 14 in 6 cases (33%). Five of them (83%) were combined with monosomy 22. Three cases (50%) were aneuploid. There was no significant correlation between trisomy 14 and histological type or prognosis.
On chromosome 14, a number of important genes have been characterized that are involved in the regulation of cell proliferation and cell death, such as FOS, the major component of the activator protein-1 (AP-1) transcription factor complex; BCL2L2 (BCL2-like2), a regulator molecule of apoptotic cell death; and TGFβ3, which controls cell proliferation and differentiation.
Conventional karyotype analysis of GCT has revealed occasional monosomy or trisomy of many other chromosomes, too (28, 29, 40). In our series we found loss of large parts of chromosome 13 and 16 and significant gains of chromosomal material of chromosome 8 in more than two cases. However, trisomy 12 and 14 and monosomy 22 seem to be most the characteristic genetic lesions of GCT.
The cytogenetic changes in GCT differ from those in epithelial ovarian cancer, which is characterized by gains at 3q, 8q, and 20q, often displaying high-level amplification. In particular, gains of chromosome 14 and loss of chromosome 22 are rarely found in ovarian carcinomas (42, 43, 44). The detailed CGH study of GCT revealed multiple smaller changes in addition; however, no high-level amplifications were revealed. As no correlation to tumor-stage or follow-up could be revealed, these aberrations probably are of secondary type.
p53 protein accumulation was found in 95% of GCT studied; however, there was no correlation with tumor stage, quantity, or type of chromosomal aberrations and survival. In particular, aberrations of chromosome 17 were only found in two cases. This suggests that accumulation of p53 protein apparently does not play such a pivotal role as has been described for ovarian carcinomas (45). Assessment of proliferative activity by Ki67 immunostaining and intensity of inhibin expression (23, 46, 47, 48, 49, 50, 51, 52, 53, 54) similarly did not provide prognostic information.
In summary, neither DNA cytometry and cytogenetics nor immunohistochemical analysis yielded prognostic information in our series of GCT. Nevertheless, the demonstration of nonrandom chromosomal aberrations may eventually lead to the identification of oncogenes and tumor suppressor genes that are pivotal for the development of GCT.
References
Hitchcock CL, Norris HJ, Khalifa MA, Wargotz ES . Flow cytometric analysis of granulosa tumors. Cancer 1989; 64: 2127–2132.
Costa MJ, Ames PF, Walls J, Roth LM . Inhibin immunohistochemistry applied to ovarian neoplasms: a novel, effective, diagnostic tool. Hum Pathol 1997; 28: 1247–1254.
Gershenson DM . Management of early ovarian cancer: germ cell and sex cord-stromal tumors. Gynecol Oncol 1994; 55: 62–72.
Evans MP, Webb MJ, Gaffey TA, Katzmann JA, Suman VJ, Hu TC . DNA ploidy of ovarian granulosa cell tumors. Cancer 1995; 75: 2295–2298.
Morikawa K, Hatabu H, Togashi K, Kataoka ML, Mori T, Konishi J . Granulosa cell tumor of the ovary: MR findings. J Comput Assist Tomogr 1997; 21: 1001–1004.
Cronje HS, Niemand I, Bam RH, Woodruff JD . Granulosa and theca cell tumors in children: A report of 17 cases and literature review. Obstet Gynecol Surv 1998; 53: 240–247.
Van den Berg-Bakker CAM, Hagemeijer A, Franken-Postma EM, Smit VTHBM, Kuppen PJK, van Ravenswaay Claasen HH, et al. Establishment and characterization of 7 ovarian carcinoma cell lines and one granulosa tumor cell line: growth features and cytogenetics. Int J Cancer 1993; 53: 613–620.
Persons DL, Hartmann LC, Herath JF, Keeney GL, Jenkins RB . Fluorescence in situ hybridization analysis of trisomy 12 in ovarian tumors. Am J Clin Pathol 1994; 102: 775–779.
Shashi V, Golden WL, von Kap-Herr C, Andersen WA, Gaffey MJ . Interphase fluorescence in situ hybridization for trisomy 12 on archival ovarian sex cord-stromal tumors. Gynecol Oncol 1994; 55: 349–354.
Cin PD, Qi H, Pauwels P, Backx C, van den Berghe H . Monosomy 22 in a fibrothecoma. Cancer Genet Cytogenet 1997; 99: 129–131.
Speleman F, Dermaut B, de Potter CR, van Gele M, van Roy N, de Paepe A, et al. Monosomy 22 in a mixed germ cell-sex cord-stromal tumor of the ovary. Genes Chromosomes Cancer 1997; 19: 192–194.
Lappöhn RE, Burger HG, Bouma J, Bangah M, Krans M, de Bruijn WA . Inhibin as a marker for granulosa-cell tumors. N Engl J Med 1989; 321: 790–793.
Matzuk MM, Finegold MJ, Su JGJ, Hsueh AJW, Bradley A . Alpha-inhibin is a tumor-suppressor gene with gonadal specificity in mice. Nature 1992; 360: 313–319.
Gurusinghe CJ, Healy DL, Jobling T, Mamers P, Burger HG . Inhibin and activin are demonstrable by immunohistochemistry in ovarian tumor tissue. Gynecol Oncol 1995; 57: 27–32.
Rishi M, Howard LN, Bratthauer GL, Tavassoli FA . Use of monoclonal antibody against human inhibin as a marker for sex cord-stromal tumors of the ovary. Am J Surg Pathol 1997; 21: 583–589.
Fuller PJ, Chu S, Jobling T, Mamers P, Healy DL, Burger HG . Inhibin subunit gene expression in ovarian cancer. Gynecol Oncol 1999; 73: 273–279.
Tanaka Y, Sasaki Y, Tachibana K, Suwa S, Tarashima K, Narkatani Y . Testicular juvenile granulosa cell tumor in an infant with X/XY mosaicism clinically diagnosed as true hermaphroditism. Am J Surg Pathol 1994; 18: 316–322.
Gordon MD, Corless C, Renshaw AA, Beckstead J . CD99, keratin and vimentin staining of sex cord-stromal tumors, normal ovary and testis. Mod Pathol 1998; 11: 769–773.
Robinson JB, Im DD, Logan L, McGuire WP, Rosenshein NB . Extraovarian granulosa cell tumor. Gynecol Oncol 1999; 74: 123–127.
Morgan DR, Brame KG . Granulosa cell tumour of the testis displaying immunoreactivity for inhibin. Br J Urol Int 1999; 83: 731–732.
Robertson DM, Cahir N, Burger HG, Mamers P, McCloud PI, Pettersson K, et al. Combined inhibin and CA125 assays in the detection of ovarian cancer. Clin Chem 1999; 45: 651–658.
Robertson DM, Cahir N, Burger HG, Mamers P, Groome N . Inhibin forms in serum from postmenopausal women with ovarian cancers. Clin Endocrinol Oxf 1999; 50: 381–386.
Gebhart JB, Roche PC, Keeney GL, Lesnick TG, Podratz KC . Assessment of inhibin and p53 in granulosa cell tumors of the ovary. Gynecol Oncol 2000; 77: 232–236.
Simon R, Bürger H, Brinkschmidt C, Böcker W, Hertle L, Terpe HJ . Chromosomal aberrations associated with progression and invasion in superficial bladder cancer. J Pathol 1995; 185: 345–351.
Hyytinen E, Visakorpi T, Kallioniemi OP, Isola J . Improved technique for analysis of formalin-fixed, paraffin-embedded tumors by fluorescence in situ hybridisation. Cytometry 1994; 16: 93–99.
Diebold J, Mösinger K, Peiro G, Pannekamp U, Kaltz C, Baretton GB, et al. 20q13 and cyclin D1 in ovarian carcinomas. Analysis by fluorescence in situ hybridization. J Pathol 2000; 190: 564–571.
Fontanelli R, Stefanon B, Raspagliesi F, Kenda R, Tomasic G, Spatti G, et al. Adult granulosa cell tumor of the ovary: a clinicopathologic study of 35 cases. Tumori 1998; 84: 60–64.
Halperin D, Visscher DW, Wallis T, Lawrence WD . Evaluation of chromosome 12 copy number in ovarian granulosa cell tumors using interphase cytogenetics. Int J Gynecol Pathol 1995; 14: 319–323.
Gorski GK, McMorrow LE, Blumstein L, Faasse D, Donaldson MH . Trisomy 14 in two cases of granulosa cell tumor of the ovary. Cancer Genet Cytogenet 1992; 60: 202–205.
Suh KS, Silverberg SG, Rhame JG, Wilkinson DS . Granulosa cell tumor of the ovary. Arch Pathol Lab Med 1990; 114: 496–501.
Christov K, Vassilev N . Flow cytometric analysis of DNA and cell proliferation in ovarian tumors. Cancer 1987; 60: 121–125.
Haba R, Miki H, Kobayashi S, Ohmori M . Combined analysis of flow cytometry and morphometry of ovarian granulosa cell tumor. Cancer 1993; 72: 3258–3262.
Jacoby AF, Young RH, Colvin R, Flotte TJ, Preffer F, Scully RE, et al. DNA content in juvenile granulosa cell tumors of the ovary: a study of early- and advanced-stage disease. Gynecol Oncol 1992; 46: 97–103.
Klemi PJ, Joensuu H, Salmi T . Prognostic value of flow cytometric DNA content analysis in granulosa cell tumor of the ovary. Cancer 1990; 65: 1189–1193.
Roush GR, El-Naggar AK, Abdul-Karim FW . Granulosa cell tumor of ovary: a clinicopathologic and flow cytometric DNA analysis. Gynecol Oncol 1995; 56: 430–434.
Diebold J, Deisenhofer I, Baretton GB, Blasenbreu S, Suchy B, Schneiderbanger K, et al. Interphase cytogenetic analysis of serous ovarian tumors of low malignant potential: comparison with serous cystadenomas and invasive serous carcinomas. Lab Invest 1996; 75: 473–485.
Fletcher JA, Gibas Z, Donovan K, Perez-Atayde A, Genest D, Morton CC, et al. Rapid communication. Ovarian granulosa-stromal cell tumors are characterized by trisomy 12. Am J Pathol 1991; 138: 515–520.
Gaffey MJ, Frierson HF Jr, Iezzoni JC, Mills SE, Clement PB, Gersell DJ, et al. Ovarian granulosa cell tumors with bizarre nuclei: an immunohistochemical analysis. Mod Pathol 1996; 9: 308–315.
Lindgren V, Waggoner S, Rotmensch J . Monosomy 22 in two ovarian granulosa cell tumors. Cancer Genet Cytogenet 1996; 89: 93–97.
Verhest A, Nedoszytko B, Noel JC, Dangou JM, Simon P, Limon J . Translocation (6;16) in a case of granulosa cell tumor of the ovary. Cancer Genet Cytogenet 1992; 60: 41–44.
Van den Berghe I, Dal Cin P, de Groef K, Michielssen P, van den Berghe H . Monosomy 22 and trisomy 14 may be early events in the tumorigenesis of adult granulosa cell tumor. Cancer Genet Cytogenet 1999; 112: 46–48.
Sonoda G, Palazzo J, du Manoir S, Godwin A, Feder M, Yakushiji M, et al. Comparative genomic hybridization detects frequent overrepresentation of chromosomal material from 3q26, 8q24, and 20q13 in human ovarian carcinomas. Genes Chromosomes Cancer 1997; 20: 320–328.
Suzuki S, Moore IID, Ginzinger D, Godfrey T, Barclay J, Powell B, et al. An approach to analysis of large-scale correlations between genome changes and clinical endpoints in ovarian cancer. Cancer Res 2000; 60: 5382–5385.
Tapper J, Bützow R, Wahlström T, Seppälä M, Knuutila S . Evidence for divergence of DNA copy number changes in serous, mucinous and endometrioid ovarian carcinomas. Br J Cancer 1997; 75: 1782–1787.
Mayr D, Pannekamp U, Baretton GB, Gropp M, Meier W, Flens MJ, et al. Immunohistochemical analysis of drug resistance-associated proteins in ovarian carcinomas. Pathol Res Pract 2000; 196: 469–475.
Ali SZ . Metastatic granulosa-cell tumor in the liver: cytopathologic findings and staining with inhibin. Diagn Cytopathol 1998; 19: 293–297.
Cooke I, O'Brien M, Charnock FM, Groome N, Ganesan TS . Inhibin as a marker for ovarian cancer. Br J Cancer 1995; 71: 1046–1050.
Costa MJ, DeRose PB, Roth LM, Brescia RJ, Zaloudek CJ, Cohen C . Immunohistochemical phenotype of ovarian granulosa cell tumors: absence of epithelial membrane antigen has diagnostic value. Hum Pathol 1994; 25: 60–66.
Flemming P, Wellmann A, Maschek H, Lang H, Georgii A . Monoclonal antibodies against inhibin represent key markers of adult granulosa cell tumors of the ovary even in their metastases. Am J Surg Pathol 1995; 19: 927–933.
Hildebrandt RH, Rouse RV, Longacre TA . Value of inhibin in the identification of granulosa cell tumors of the ovary. Hum Pathol 1997; 28: 1387–1394.
Jobling T, Mamers P, Healy DL, MacLachlan V, Burger HG, Quinn M, et al. A prospective study of inhibin in granulosa cell tumors of the ovary. Gynecol Oncol 1994; 55: 285–289.
Kommoss F, Oliva E, Bhan AK, Young RH, Scully RE . Inhibin as a marker for granulosa-cell tumors. Mod Pathol 1998; 11: 656–664.
Yamato M, Minami S, Nakano R, Kobayashi M . Immunohistochemical localization of inhibin/activin subunits in human ovarian follicles during the menstrual cycle. J Clin Endocrinol Metab 1992; 74: 989–993.
Yamashita K, Yamoto M, Shikone T, Minami S, Imai M, Nichimori K, et al. Production of inhibin A and inhibin B in human ovarian sex cord stromal tumors. Am J Obstet Gynecol 1997; 177: 1450–1457.
Acknowledgements
The authors thank Astrid Hirschmann, Beate Luthardt, and Nathalie Peterka for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mayr, D., Kaltz-Wittmer, C., Arbogast, S. et al. Characteristic Pattern of Genetic Aberrations in Ovarian Granulosa Cell Tumors. Mod Pathol 15, 951–957 (2002). https://doi.org/10.1097/01.MP.0000024290.55261.14
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000024290.55261.14
Keywords
This article is cited by
-
Immunohistochemical markers of prognosis in adult granulosa cell tumors of the ovary – a review
Journal of Ovarian Research (2023)
-
Keimstrang-Stromatumoren des Ovars
Der Pathologe (2019)
-
KMT2D/MLL2 inactivation is associated with recurrence in adult-type granulosa cell tumors of the ovary
Nature Communications (2018)
-
Combined comparative genomic hybridization and transcriptomic analyses of ovarian granulosa cell tumors point to novel candidate driver genes
BMC Cancer (2015)
-
Diagnose und Differenzialdiagnose des Granulosazelltumors
Der Pathologe (2007)
