
Abstract
Primary anaplastic large-cell lymphoma is a rare malignancy in the lung. Anaplastic large-cell lymphoma characteristically involves the lymph nodes or skin, with few reports from other sites. We studied the clinical and pathologic features of five cases of anaplastic large-cell lymphoma limited to the lungs. The patients were three women and two men aged 27 to 66 years (mean, 44.6 y) The tumors ranged in size from 1.1 to 5 cm. All patients were CD 30 (Ki-1) positive and CD 15 (LeuM-1) negative. Epithelial membrane antigen immunoreactivity was seen in two patients. Epstein-Barr virus was not detected by immunohistochemistry (four patients tested) or by polymerase chain reaction studies (three patients tested). The immunophenotypes were T cell (n = 3) and null (n = 2). Gene rearrangement studies supported the immunophenotypic findings. One patient who had underlying HIV infection died of infectious complications. One patient died at 6 months. Two patients developed recurrent disease and are alive after 42 and 51 months of follow-up. The remaining patient is alive at 8 years of follow-up without evidence of disease. ALCL can mimic metastatic or primary carcinoma and should be considered in the differential diagnosis of large cell neoplasms of the lung.
Similar content being viewed by others


INTRODUCTION
First described by Stein and colleagues in 1985 (1), anaplastic large-cell lymphoma (ALCL) is characterized by pleomorphic large cells and consistent expression of CD30/Ki-1. ALCL may have a prominent sinusoidal growth pattern (in lymph nodes) that may mimic metastatic carcinoma. ALCL has a propensity to involve extranodal sites, particularly skin (2, 3, 4). However, presentation in the lung is rare, reported in only two patients to date (3, 5). In this study, we describe the clinicopathologic features of five additional cases of primary ALCL of the lung.
MATERIALS AND METHODS
Source Materials
We retrieved from the files (1970–1996) of the Armed Forces Institute of Pathology all cases coded as ALCL, lymphocyte-depleted Hodgkin's disease (HD), and malignant histiocytosis/true histiocytic lymphoma primary in the lung. A total of 28 cases were retrieved. Using the histomorphologic and immunophenotypic criteria of Stein et al. (1), five cases were diagnosed as ALCL. In accordance with the Revised European–American Lymphoma
Classification system, cases of B-cell lymphoma that had ALCL morphology and immunoreactivity for CD 30 were excluded (6). Each patient had hematoxylin-eosin–stained slides and a complete clinical history available. All patients had paraffin-embedded tissue or unstained, positively charged slides available for immunophenotypic analysis. To be included in this study, the patients were required to present with a pulmonary lesion and to have no clinical, radiographic, or pathologic evidence of extrapulmonary involvement previously, concurrently, or within 3 months after diagnosis. This is in keeping with the criteria for primary pulmonary lymphoma proposed by L' Hoste et al. (7). Follow-up consisted of questionnaires prepared in accordance with the Armed Forces Institute of Pathology Human Use Committee and the statutes of the Privacy Act.
Immunohistochemistry
Immunohistochemical studies were performed according to the avidin–biotin complex method of Hsu (8). The antibodies included CD45RB (leukocyte common antigen; mouse monoclonal, 1:100 dilution; DAKO, Carpinteria, CA); CD20 (L26; mouse monoclonal, 1:200 dilution; DAKO); CD45RO (UCHL-1; mouse monoclonal, 1:200 dilution; DAKO); CD3 (rabbit polyclonal 1:50 dilution; DAKO); CD43 (MT1; mouse monoclonal, 1:50 dilution; Biotest, Denville, NJ); cytokeratin (AE1/AE3; mouse monoclonal, 1:400 dilution; Boehringer Mannheim, Indianapolis, IN); CD15 (LeuM1; mouse monoclonal, 1:100 dilution; Becton-Dickinson, San Jose, CA); CD30 (Ki-1; mouse monoclonal,1:100 dilution; DAKO); epithelial membrane antigen (mouse monoclonal, 1:100 dilution; DAKO); and latent membrane protein-1 (mouse monoclonal, 1:50 dilution; DAKO). The antibodies for cytokeratin, CD15, CD30, and latent membrane protein required predigestion for 3 minutes with 0.05 Protease VIII (Sigma Chemical Co, St. Louis, MO) in 0.1 m phosphate buffer at a pH of 7.6 at 37 °C. CD3 required a 10-minute predigestion for optimal results. Insufficient material remained for the performance of immunohistochemical stains to assess the presence of the NPM-ALK fusion protein (ALK-1).
Molecular Diagnostic Studies
Polymerase chain reaction (PCR) studies for rearrangement status of the immunoglobulin heavy chain gene, T-cell receptor (TCR)-β and -γ chain genes; for the detection of Epstein-Barr virus (EBV); and for the t(2;5) translocation transcript were performed as described elsewhere (9, 10, 11, 12). Briefly, DNA- and RNA-containing lysates were extracted from formalin-fixed, paraffin-embedded tissue sections by methods described in Wright et al. (9) and Krafft et al. (10) in the three cases for which blocks were available. PCR for immunoglobulin heavy chain and TCR rearrangement status and EBV detection were performed as described in Abruzzo et al. (11). PCR for the t(2;5) translocation was performed as described in Morris et al. (12).
RESULTS
Clinical Features
There were three women and two men ranging in age from 27–66 years (mean, 44.6 y;Table 1). Two of the five patients were known to be smokers. Four patients presented with symptoms related to their tumors, including fever, cough, night sweats, and weight loss. One patient (Patient 4) presented with symptoms related to his underlying HIV infection. This patient also had a history of prostate adenocarcinoma. The left lung alone was involved in two patients and the right lung alone in one patient. Patient 4 presented with multiple bilateral nodules; the maximum diameter of any nodule was 1.1 cm. Patient 5 presented with an intratracheal mass with extension into the main stem bronchi, mainly on the right. The tumor presented radiographically as a solitary nodule or mass in four cases, one of which appeared cystic. Clinical and radiological staging revealed no evidence of mediastinal or extrathoracic disease in any of the patients.
Of the five patients, one (Patient 4) with associated HIV infection died with sepsis 21 days after presentation. The remaining four patients were treated with surgery and multiagent chemotherapy; three patients also underwent radiotherapy. Patient 1 achieved clinical remission and is without evidence of disease at follow-up of 8 years. Two questionable nodules found in the left lower lobe on computed tomography scan during initial staging were presumed to be tumor and were also treated with local radiotherapy. Patient 2 had remission lasting 38 months. At that point, a routine follow-up computed tomography scan showed local recurrence that was confirmed by biopsy. Additional chemotherapy, followed by stem cell transplant, effected remission. The patient is now without evidence of disease at 51 months after original diagnosis. After 4 months, Patient 3 developed multiple erythematous skin lesions and a soft-tissue thigh mass. Biopsy of the thigh mass confirmed that it represented ALCL. A biopsy of one of the skin lesions reportedly showed similar histology. The soft tissue mass grew rapidly and was resected 2 months later. This patient was alive 42 months after original diagnosis but was subsequently lost to follow-up. The patient had presented in a remote area, and it is unclear whether he had any evidence of disease at the time of that last follow-up. The patient in Case 5 had a difficult clinical course, with airway obstructions, and died 6 months into the follow-up period.
Pathologic Findings
The neoplasms were unifocal in four patients (Patients 1–3 and 5) and multifocal in Patient 4. They varied from 1.1 to 5.0 cm in greatest diameter. One tumor was cystic, and the others were solid. Two tumors (Patients 3 and 5) had intra-airway (bronchial or tracheal) projections (Figure 1), and the others were well-circumscribed masses (Table 1). The tumor in Patient 2 was described as a tan-yellow cystic mass (Figure 2) with extensive necrosis; the tumors in the remaining four patients were homogeneously tan. Pneumonectomy was performed in Patient 3. Hilar lymph nodes resected in that case showed no tumor.

Intrabronchial polypoid growth of lymphoma. Case 3.

Lymphomatous tumor nodule with extensive necrosis en masse. Case 2.
On low-power microscopic examination, all patients exhibited obliteration of the normal alveolar architecture. Patient 1 had areas of a nodular growth pattern and, along with Patient 2, demonstrated intra-alveolar spread of tumor cells, producing a “tumoral pneumonia” (Fig. 3). Patient 2 had closely scattered nodules with cyst formation in some but lacked the nodular growth seen in Patient 1. Patients 3 and 5 had intraluminal polypoid masses that partially occluded large airways. Patient 4 exhibited well-circumscribed nodules separated by uninvolved lung parenchyma. A lymphangitic distribution was not observed in any of the cases.

Neoplastic infiltrate of alveolar airspaces produces a tumoral pneumonia. The alveolar septa are greatly widened by neoplastic and reactive elements. The airspaces are filled predominantly with tumor cells. Case 1.
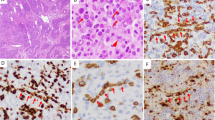
Three cases (1, 4, and 5) consisted of relatively monomorphic large cells with vesicular nuclei, multiple inconspicuous nucleoli, and pale gray cytoplasm (Fig. 4). Multinucleated giant cells were not a prominent feature in these two cases. In Patients 2 and 3, the tumors consisted of large cells with pleomorphic nuclei, one or more prominent nucleoli, and abundant eosinophilic cytoplasm (Fig. 5). Multinucleated giant cells, some with Reed-Sternberg–like features, were frequently observed. In all patients, variable numbers of neutrophils and small lymphocytes were admixed with the malignant cells; eosinophils were prominent in Patients 1 and 2. Necrosis was observed in all patients and was particularly extensive in Patients 2 and 3. Vascular invasion by malignant cell infiltrates was noted in Patients 4 and 5.

Pale monomorphous tumor cells. The alveolar septa are not appreciably widened in this field. Case 1.

Large pleomorphic neoplastic cells with prominent nucleoli and eosinophilic cytoplasm. Case 3.
Secondary inflammatory changes in the adjacent lung parenchyma were noted in three cases. Acute bronchopneumonia combined with obstructive pneumonia with distension of the alveoli by foamy histiocytes was seen in two patients (Patients 1 and 2). Organizing pneumonia was noted in Patient 1. Foci of a reactive perivascular lymphocytic infiltrate were noted in areas uninvolved by neoplasm in Patients 1 and 2. A desquamative interstitial pneumonia (DIP)–like reaction with numerous intra-alveolar macrophages was seen in Patient 2. Patient 5 exhibited squamous metaplasia of an entrapped bronchial gland.
Immunohistochemistry
In all cases, the tumor cells showed strong membrane and Golgi immunoreactivity with CD30 (Fig. 6) and were negative for keratin and CD15 (Table 2). CD45RB (LCA) was positive in three of five cases; two of five cases showed membrane and Golgi staining for epithelial membrane antigen. Three cases demonstrated a T-cell immunophenotype (in Patients 1, 3, and 4). The remaining two cases failed to stain with markers of B- or T-cell differentiation. In the four cases for which material was available, the immunohistochemical stain for EBV (latent membrane protein) was negative (Table 3).

CD 30 immunohistochemical stain demonstrating immunoreactivity in the Golgi zone and at the cytoplasmic membrane of the neoplastic cells. Case 4.
Molecular Findings
In three cases (Patients 1, 3, and 4), PCR for immunoglobulin heavy chain, TCR-β, and TCR-γ gene rearrangements were performed (Table 3). For two of the three cases with a T-cell immunophenotype (Patients 3 and 4), dominant monoclonal rearrangements of the TCR-β and -γ chains were demonstrated. The remaining case (Patient 1) showed no monoclonal rearrangements of either the immunoglobulin heavy chain or T-cell receptor genes. Neither the t(2;5) chromosomal translocation nor the presence of EBV genomes was detected (by PCR) in any of the cases tested.
DISCUSSION
Non-Hodgkin's lymphoma arising in the lung is rare, accounting for approximately 3.6% of extranodal lymphomas (13) and for only 0.3% of primary lung neoplasms (14). The majority of lymphomas are low-grade B-cell type and are believed to be derived from bronchus-associated lymphoid tissue (BALT; 15). These low-grade lymphomas show a peak occurrence in the 6th decade, are frequently asymptomatic at presentation, and have a relatively indolent clinical course (16, 17). Approximately 5% of pulmonary lymphomas are follicular center cell, and up to 20% are high-grade neoplasms (15, 16). In the high-grade category, the majority are diffuse, large B-cell lymphomas with centroblastic/immunoblastic morphology (Revised European–American Lymphoma Classification; 6). A small percentage of cases are associated with low-grade MALT lymphoma and are thought to represent a high-grade transformation. Unlike low-grade B-cell lymphomas, high-grade lymphomas typically present with respiratory/constitutional symptoms and have a poorer prognosis (16).
ALCL is a relatively recently described type of non-Hodgkin's lymphoma characterized by strong immunoreactivity with the monoclonal antibodies Ki-1/Ber H2 (CD30; 1). These lymphomas usually express a T-cell phenotype. Some cases do not exhibit a specific phenotype and are often referred to as the null phenotype. ALCLs generally behave in an aggressive manner (6, 18). Extranodal disease is common in ALCL, occurring in 25–65% of patients (2) on skin, by far the most commonly reported extranodal site. Although lung involvement may occur as a result of dissemination in up to 12% of cases (3), primary pulmonary ALCL has only been reported in rare cases (3, 5).
Patients with ALCL have a bimodal age distribution at presentation, with increased incidence in children (18). Chott et al. (3) reported a median age of 50 years. In the current series, all patients were adults, with the mean age being 44.6 years and the median age, 34 years; they are therefore somewhat younger than patients with other primary pulmonary lymphoma, who tend to present in the 6th or 7th decade (15, 17). Although smoking has been strongly correlated with pulmonary Langerhan's cell histiocytosis (19), it has not been assessed as a possible risk factor for other pulmonary hematopoietic lesions except for the report of Cordier et al. (17),who recorded 27 of 66 pulmonary lymphoma patients with a smoking history. About half of patients with primary pulmonary low-grade B-cell lymphomas are asymptomatic at presentation, whereas those with higher grade lesions tend to present with respiratory or constitutional symptoms (15, 16, 17). The presence of constitutional symptoms has been reported as an unfavorable prognostic sign, in whose absence even patients with high-grade lesions may do well (16). Overall, the 5-year survival rate for high-grade lymphomas has been reported to be 60% (16). In our series with up to 8 years of follow-up, one death was attributed to sepsis and one to disease. The presentation of an intraluminal large airway mass, seen in two patients, is unusual for hematologic lesions of the lung (20). However, one of our patients presented with a cavitating mass, a finding occasionally seen in high-grade pulmonary lymphomas (17, 21).
The low-power histologic features of our cases are similar to those previously reported in studies of high-grade primary pulmonary lymphoma (15, 16). In this series, all patients showed at least focal obliteration of the lung architecture, with two patients showing an associated intra-alveolar spread of tumor cells. The case in Patient 1, in particular, had the appearance of a neoplastic pneumonia. In comparing low-grade with high-grade lymphomas, Li et al. (16) described intra-alveolar spread, necrosis, and vascular infiltration by tumor cells as distinctive features of the high-grade neoplasms. Parenthetically, necrosis was present in each of the current cases and prominently so in two of five cases (Patients 2 and 3). Vascular invasion was observed in two of five cases (Patients 4 and 5) in this series.
ALCL comprises a cytomorphologically heterogeneous category. Since the first reports in 1985, a number of morphologic variants have been described. The most frequent variant is called the common type (3, 22). Cellular morphology within this type varies from monomorphic to pleomorphic and from pale cell to basophilic cell (3, 23). Other variants include lymphohistiocytic (22), giant cell (22), sarcomatoid (24),and small cell (25). In this series, three cases exhibited monomorphic cytology (in Patients 1, 4, and 5), and two cases exhibited pleomorphic cytology (in Patients 2 and 3).
The immunophenotypic findings in our cases are similar to those of previous reports of ALCL in other sites, with three of five (60%) expressing a T-cell immunophenotype and two of five (40%), null (18). None of the cases in this series showed an association with EBV by either immunohistochemical staining (latent membrane protein) or PCR techniques. Low rates of EBV (16–18%) have been reported in ALCL in previous studies from other sites (26). There appears to be an infrequent association between ALCL and EBV as compared with HD (40–80%) (27).
In the three cases for which sufficient material allowed for a PCR-based assay for the t(2;5) translocation, no evidence for the translocation was found. Tumors that express ALK-1 protein product of the t(2;5) translocation are reported to have a more favorable prognosis and to represent a distinct clinicopathologic entity from those that do not (28).
Primary ALCL of the lung has been reported in two cases that were included in larger series of ALCL or HIV-associated ALCL (3, 5, 29). The HIV-associated case was of T-cell phenotype (5), and the other was classified as null type (3). A third case included in a series reported by Nosari et al. (29) was also HIV-related and was of B-cell phenotype. CD 30 immunoreactive B-cell lymphomas are not considered in the ALCL category in the Revised European–American Lymphoma Classification, however (6). Each neoplasm behaved aggressively, with survival of less than 3 months. In the current series, Patient 4, with an underlying HIV infection, had an aggressive T-cell ALCL and died of sepsis 21 days after diagnosis. Patient 5 had an unresectable tumor of null phenotype and died at 6 months. The remaining three patients showed a more favorable clinical course and were alive at follow-ups ranging from 42 to 100 months. In comparison, pleomorphic carcinoma of the lung has a 5-year survival rate of 11.8% (30).
The principal differential diagnoses of pulmonary ALCL include primary or metastatic carcinoma, HD, and lymphomatoid granulomatosis (LYG). Considering that carcinoma accounts for greater than 90% of invasive lung malignancies, excluding this possibility is of prognostic and therapeutic relevance. Similar to the case of ALCL, certain histologic subtypes of lung carcinoma, particularly the pleomorphic/giant cell subtypes, exhibit bizarre cytomorphology and multinucleated giant cells and may have a mixed background inflammation. Pale, vacuolated cells and entrapped benign epithelial elements in cases of ALCL of lung (as in Patient 5) may mislead toward a diagnosis of carcinoma. Finally, ALCL may show overlapping immunohistochemical findings with pleomorphic carcinoma. Up to one third of ALCL cases lack CD45RB (LCA) immunoreactivity in paraffin sections, whereas reactivity for epithelial membrane antigen has been reported in the majority of cases (31). Indeed, in rare cases of ALCL, cytokeratin has been detected in both frozen and paraffin sections (32). In contrast, pleomorphic carcinoma typically shows immunoreactivity with cytokeratin and/or epithelial membrane antigen, does not express CD45RB or CD30, and lacks lineage-specific antigens for B and T cells. These findings highlight the importance of evaluating a complete battery of hematopoietic and epithelial markers in differentiating these two pleomorphic neoplasms.
Primary pulmonary HD is rare; most cases involving the lung parenchyma occur as a result of extension from mediastinal nodal sites or as multinodular lymphangitic metastasis in cases with established HD elsewhere in the body (33, 34). The most commonly reported subtypes involving the lung are nodular sclerosis and mixed cellularity, with only rare occurrences of the lymphocyte depletion and lymphocyte predominance subtypes. ALCL resembles HD in terms of the presence of occasional Reed-Sternberg–like cells and of immunoreactivity with CD30. In most cases, HD can be readily distinguished from ALCL by the lack of a cohesive growth pattern and the low ratio of neoplastic cells in comparison to reactive background cells. However, several unusual variants of nodular sclerosis HD, the so-called syncytial variant and the Grade II subtype proposed by the British National Lymphoma Investigation, are characterized by increased neoplastic cellularity, pleomorphism, and lymphoid depletion (35). These entities can be very difficult to distinguish from ALCL, especially in small biopsy specimens. The nodular growth pattern observed in Patient 1 and the marked eosinophilia in both Patients 1 and 2 may suggest a diagnosis of HD. Eosinophil-rich and neutrophil-rich variants of ALCL have been described elsewhere (36). Although the case in Patient 1 showed sclerotic bands, the monomorphic appearance of the large cells, lack of staining with CD15, and positivity for CD45RO and CD43 (MT1) were more supportive of a diagnosis of ALCL. The case in Patient 2 demonstrated positive staining with CD45RB, which is not characteristic of HD.
The necrosis and vascular invasion demonstrated by several cases of ALCL superficially resembled the histologic features of LYG. However, a number of morphologic and immunophenotypic features allow reliable differentiation of these two entities. Although foci of angioinvasion were seen in two patients, the striking angiocentricity that characterizes LYG was not seen. Whereas ALCL is a cytologically malignant large-cell proliferation, LYG shows a polymorphous cellular infiltrate with varying but often small to moderate numbers of atypical large monotypic B cells in a background of small reactive T lymphocytes and histiocytes (37). Furthermore, in the majority of cases of LYG, the large B-cells contain EBV genome (37). In contrast, most cases of ALCL are of T-cell phenotype, and EBV has been detected only in a minority of cases (26). Of interest, however, Myers (38) described a subset of angiocentric T-cell lymphomas with LYG morphology that lacked evidence for the presence of the EBV genome.
Although a rare tumor, primary lung ALCL should be considered in the differential diagnosis of other large cell neoplasms of the lung, such as large-cell and pleomorphic carcinomas of the lung, HD, and LYG. Follow-up data to date suggest a favorable prognosis for low-stage, resectable ALCL of the lung.
References
Stein H, Mason DY, Gerdes J, O'Connor N, Wainscoat J, Pallesen G, et al. The expression of the Hodgkin's disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985; 66: 848–858.
Greer JP, Kinney MC, Collins RD, Salhany KE, Wolff SN, Hainsworth JD, et al. Clinical features of 31 patients with Ki-1 anaplastic large-cell lymphoma. J Clin Oncol 1991; 9: 539–547.
Chott A, Kaserer K, Augustin I, Vesely M, Heinz R, Oehlinger W, et al. Ki-1-positive large cell lymphoma. A clinicopathologic study of 41 cases. Am J Surg Pathol 1990; 14: 439–448.
Kadin ME . The spectrum of Ki-1+ cutaneous lymphomas. Curr Probl Dermatol 1990; 19: 132–143.
Chadburn A, Cesarman E, Jagirdar J, Subar M, Mir RN, Knowles DM . CD30 (Ki-1) positive anaplastic large cell lymphomas in individuals infected with the human immunodeficiency virus. Cancer 1993; 72: 3078–3090.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, et al. Lymphoma classification proposal: clarification. Blood 1995; 85: 857–860.
L'Hoste RJ Jr, Filippa DA, Lieberman PH, Bretsky S . Primary pulmonary lymphomas. A clinicopathologic analysis of 36 cases. Cancer 1984; 54: 1397–1406.
Hsu SM . Immunohistochemistry. Methods Enzymol 1990; 184: 357–363.
Wright CF, Reid AH, Tsai MM, Ventre KM, Murari PJ, Frizzera G . Detection of Epstein-Barr virus sequences in Hodgkin's disease by the polymerase chain reaction. Am J Pathol 1991; 139: 393–398.
Krafft A, Duncan B, Bijwaard K, Taubenberger J, Lichy J . Optimization of the isolation and amplification of RNA from formalin-fixed, paraffin-embedded tissue: the AFIP experience and literature review. Mol Diagn 1997; 2: 217–230.
Abruzzo LV, Griffith LM, Nandedkar M, Aguilera NS, Taubenberger JK, Raffeld M, et al. Histologically discordant lymphomas with B-cell and T-cell components. Am J Clin Pathol 1997; 108: 316–323.
Morris SW, Kirstein MN, Valentine MB, Dittmer K, Shapiro DN, Look AT, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma [letter]. Science 1995; 267: 316–317.
Freeman C, Berg JW, Cutler SJ . Occurrence and prognosis of extranodal lymphomas. Cancer 1972; 29: 252–260.
Miller D, Allen M . Rare pulmonary neoplasms. Mayo Clin Proc 1993; 68: 492–498.
Koss MN, Hochholzer L, Nichols PW, Wehunt WD, Lazarus AA . Primary non-Hodgkin's lymphoma and pseudolymphoma of lung: a study of 161 patients. Hum Pathol 1983; 14: 1024–1038.
Li G, Hansmann ML, Zwingers T, Lennert K . Primary lymphomas of the lung: morphological, immunohistochemical and clinical features. Histopathology 1990; 16: 519–531.
Cordier JF, Chailleux E, Lauque D, Reynaud-Gaubert M, Dietemann-Molard A, Dalphin JC, et al. Primary pulmonary lymphomas: a clinical study of 70 cases in nonimmunocompromised patients. Chest 1993; 103: 201–208.
Kadin ME . Primary Ki-1-positive anaplastic large-cell lymphoma: a distinct clinicopathologic entity. Ann Oncol 1994; 5 (1 Suppl):25–30.
Hance AJ, Basset F, Saumon G, Danel C, Valeyre D, Battesti JP, et al. Smoking and interstitial lung disease. The effect of cigarette smoking on the incidence of pulmonary histiocytosis X and sarcoidosis. Ann N Y Acad Sci 1986; 465: 643–656.
Samuels M, Howe C, Dodd G Jr, Fuller L, Shullenberger C, Leary W . Endobronchial malignant lymphoma. report of five cases in adults. AJR Am J Roentgenol 1961; 85: 87–95.
Jackson SA, Tung KT, Mead GM . Multiple cavitating pulmonary lesions in non-Hodgkin's lymphoma. Clin Radiol 1994; 49: 883–885.
Pileri SA, Piccaluga A, Poggi S, Sabattini E, Piccaluga PP, De Vivo A, et al. Anaplastic large cell lymphoma: update of findings. Leuk Lymphoma 1995; 18: 17–25.
Chan JK, Ng CS, Hui PK, Leung TW, Lo ES, Lau WH, et al. Anaplastic large cell Ki-1 lymphoma. Delineation of two morphological types. Histopathology 1989; 15: 11–34.
Chan JK, Buchanan R, Fletcher CD . Sarcomatoid variant of anaplastic large-cell Ki-1 lymphoma. Am J Surg Pathol 1990; 14: 983–988.
Kinney MC, Collins RD, Greer JP, Whitlock J, Sioutos N, Kadin ME . A small-cell-predominant variant of primary Ki-1 (CD30)+ T-cell lymphoma. Am J Surg Pathol 1993; 17: 859–868.
Lopategui JR, Gaffey MJ, Chan JK, Frierson HF, Sun LH, Bellafiore FJ, et al. Infrequent association of Epstein-Barr virus with CD30-positive anaplastic large cell lymphomas from American and Asian patients. Am J Surg Pathol 1995; 19: 42–49.
Pinkus GS, Lones M, Shintaku IP, Said JW . Immunohistochemical detection of Epstein-Barr virus-encoded latent membrane protein in Reed-Sternberg cells and variants of Hodgkin's disease. Mod Pathol 1994; 7: 454–461.
Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ, et al. ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 2000; 91: 2076–2084.
Nosari A, Cantoni S, Oreste P, Schiantarelli C, Landonio G, Alexiadis S, et al. Anaplastic large cell (CD30/Ki-1+) lymphoma in HIV+ patients: clinical and pathological findings in a group of ten patients. Br J Haematol 1996; 95: 508–512.
Travis WD, Travis LB, Devesa SS . Lung cancer [published erratum appears in Cancer 1995 Jun 15; 75(12): 2979]. Cancer 1995; 75 (1 Suppl):191–202.
Delsol G, Al Saati T, Gatter KC, Gerdes J, Schwarting R, Caveriviere P, et al. Coexpression of epithelial membrane antigen (EMA), Ki-1, and interleukin-2 receptor by anaplastic large cell lymphomas. Diagnostic value in so-called malignant histiocytosis. Am J Pathol 1988; 130: 59–70.
Frierson HF Jr, Bellafiore FJ, Gaffey MJ, McCary WS, Innes DJ Jr, Williams ME . Cytokeratin in anaplastic large cell lymphoma. Mod Pathol 1994; 7: 317–321.
Yousem SA, Weiss LM, Colby TV . Primary pulmonary Hodgkin's disease. A clinicopathologic study of 15 cases. Cancer 1986; 57: 1217–1224.
Radin AI . Primary pulmonary Hodgkin's disease. Cancer 1990; 65: 550–563.
MacLennan KA, Bennett MH, Tu A, Hudson BV, Easterling MJ, Hudson GV, et al. Relationship of histopathologic features to survival and relapse in nodular sclerosing Hodgkin's disease. A study of 1659 patients. Cancer 1989; 64: 1686–1693.
McCluggage WG, Walsh MY, Bharucha H . Anaplastic large cell lymphoma with extensive eosinophilic or neutrophilic infiltration. Histopathology 1998; 32: 110–115.
Guinee D Jr, Jaffe E, Kingma D, Fishback N, Wallberg K, Krishnan J, et al. Pulmonary lymphomatoid granulomatosis. Evidence for a proliferation of Epstein-Barr virus infected B-lymphocytes with a prominent T-cell component and vasculitis. Am J Surg Pathol 1994; 18: 753–764.
Myers JL . Lymphomatoid granulomatosis: past, present,…future? Mayo Clin Proc 1990; 65: 274–278.
Acknowledgements
The authors would like to express their gratitude for the photographic assistance of Mr. Luther Duckett (medical microphotographer) and Ms. Robin-Anne V. Ferris, M.F.S. (section chief) of the photography section, Armed Forces Institute of Pathology.
Author information
Authors and Affiliations
Additional information
The findings and opinions presented in this paper do not necessarily represent official positions of the Department of Defense or of the Army or Air Force.
Address reprint requests to: Department of Pulmonary and Mediastinal Pathology Armed Forces Institute of Pathology, Washington, DC 20306-6000; fax: 202-782-3075.
Rights and permissions
About this article
Cite this article
Rush, W., Andriko, J., Taubenberger, J. et al. Primary Anaplastic Large Cell Lymphoma of the Lung: A Clinicopathologic Study of Five Patients. Mod Pathol 13, 1285–1292 (2000). https://doi.org/10.1038/modpathol.3880235
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880235
Keywords
This article is cited by
-
p40 (ΔNp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma
Modern Pathology (2012)
