
Abstract
Two problems were the focus of this study. (1) Is precancer and/or invasive cancer of the human cervix a poly- or monoclonal proliferation of neoplastic cells? (2) Are simultaneously present precancers and cancers of the cervix clonally related, or do they arise independently? Microdissection of 37 neoplastic lesions with different degrees of histologic severity in 22 patients followed by polymerase chain reaction–based analysis of X-chromosome inactivation was used as a principal method. Invasive cancers were interpreted as monoclonal because samples invariably showed monoclonal signals. In two thirds of these cases, simultaneously present precursors had the identical X-chromosome inactivation pattern, but in one third the pattern was different. Polyclonality was seen in a subgroup of precursors, where there was no simultaneous presence of invasive cancer. In contrast, when invasive cancer was present, no precursor signaled polyclonality. Data taken together indicate that the pathogenesis of cervical cancer is probably even more complicated than that of other cancers involving selection of subclones from originally polyclonal precursors and possibility of coexistence of precursors of different monoclonal composition. The study also observed that a large field of normal cervical squamous epithelium (approximately 500 basal squamous epithelial cells) with nonrandom X-chromosome inactivation was present. It remains to be further investigated whether this phenomenon represents an embryologic lyonization pattern of X-chromosome inactivation or postembryologic clonal expansion of submorphologically transformed cells.
Similar content being viewed by others


INTRODUCTION
Monoclonality (origin from a single transformed cell) has been accepted as a fundamental feature of human cancer (1, 2). This concept largely rests on experimental observations of cancers in invasive stage. Clonal dominance in late stages (3) does not, however, rule out a polyclonal origin because selection may have caused loss of less competitive clones (4). A way to resolve this issue is to examine the clonal composition of premalignant or early malignant lesions (4, 5, 6). We chose the X-chromosome inactivation marker as our tool. It is presumed to be neutral (i.e., of no selective value) and subject to permanent inactivation by a random process (7, 8).
Cervical neoplasia, in which different precursors of invasive cancer are common and morphologically identified with ease, provides a good model for studies of clonality (9). In the vast majority of precancers, human papillomavirus (HPV) DNA is found (10). It is not known, however, whether the precancers reflect polyclonal proliferation of a number of simultaneously infected cells or clonal outgrowth of progeny of one cell that, after interaction with HPV plus other unknown factors, has “transformed” to precancer and expanded by proliferation. The second problem addressed is the genetic relation between precancer and invasive cervical cancer. By examination of simultaneously present lesions, we wanted to establish whether they had an independent origin or were genetically linked.
Polymerase chain reaction (PCR)-based strategy for X-chromosome inactivation analysis (11) makes it possible to analyze archival tissues (12). In a previous study, we analyzed the clonality of cervical cancers by combining PCR-based X-chromosome inactivation analysis of the X-linked androgen receptor gene with microdissection from histologic sections (13). In this study, we have used our modified method to determine the clonality of cervical precancers and their somatic genetic relation to coexisting invasive cancers.
MATERIALS AND METHODS
Case Selection
Twenty-two cases of multiple cervical neoplastic lesions, heterozygotic at the androgen receptor gene locus, were collected for analysis. Multiple lesions were identified as topographically discontinuous entities from individual cases. All cases were collected from the Department of Genetics and Pathology, Uppsala University Hospital, Sweden, and Department of Pathology of the Gynecology Hospital, Helsinki University, Finland. Tissues had been stored for 2 to 4 years as formaldehyde-fixed, paraffin-embedded specimens.
Microdissection
Five 5-μm sections were cut from each tissue block. Slides were deparaffinized in xylene for 5 min twice and processed through increasingly hydrated alcohol. Each section was stained with one or two drops of methylene blue for 7 seconds, washed with running distilled water for a few minutes, and then dried at room temperature. Neoplastic areas or normal squamous epithelium with or without underlying stroma were dissected directly from the stained slides under the microscope by fine surgical scalpels. The amount of neoplastic area microdissected varied from lesion to lesion according to the size of lesion for clear separation from surrounding non-neoplastic tissues. However, at least 5000 cells from each lesion were ensured for analysis. Approximately 500 cells per sample were microdissected from normal cervical epithelia to determine roughly the minimal size of “tiles” of X-chromosome inactivation in cervical squamous epithelium. Precautions to prevent cross-contamination during microdissection included changing the surgical instruments from section to section and rinsing the slides individually.
DNA Preparation
Microdissected neoplastic and normal tissue was transferred to 1.5 mL Eppendorff tubes with 200 μl 0.01 m Tris (pH 8.0), 0.01 m EDTA, and 1% sodium dodecyl sulfate (SDS) with 500 μg/mL proteinase K. The samples were incubated overnight at 56° C. After completion of proteinase-K digestion, the samples were precipitated with 50 μl saturated NaCl (approximately 6 m) followed by centrifugation at 2000 g for 10 min. Supernatants were then transferred to new 1.5 mL Eppendorff tubes, and DNA was precipitated with 2 volumes of absolute ethanol at −70° C for at least 2 h. The samples were centrifuged in 12,000 g for 20 min. Precipitated DNA was washed with 80% alcohol and resuspended in 30 μl distilled water.
Restriction Enzyme Treatment
Ten microliters of each DNA sample were used for a 20-μl reaction mixture containing 20 U HhaI (Pharmacia, Uppsala, Sweden) and incubated at 37° C overnight. The reaction was stopped by heating at 70° C for 20 min. Another 20-μl mixture was prepared from the DNA sample and treated in exactly the same way except for omission of HhaI.
PCR Amplification
The principal advantage of the androgen receptor gene as X-chromosome inactivation marker has been described (14). Primers for amplification were from DNA Technology (Aarhus, Denmark). Primer sequences were 5′-TCC AGA ATC TGT TCC AGA GCG TGC-3′ and 5′-GCT GTG AAG GTT GCT GTT CCT CAT-3′. Ten microliters from each DNA sample were added to 50 μl PCR reaction mixture containing 1 μm of each primer; standard PCR reaction buffer (Perkin Elmer Cetus, Norwalk, CT); 200 μm ATP, GTP, CTP, TTP, respectively; and 1 U Taq polymerase (Perkin Elmer Cetus). Forty cycles of amplification were carried out using cycling parameters of 95° C for 30 seconds, 68° C for 30 seconds, and 72° C for 60 seconds with initial 95° C for 3 min. Three microliters of PCR products were mixed with an equal volume of gel loading buffer containing 95% formamide, 0.25% bromophenol, and 0.25% xylene cyanol, loaded onto 7.2 m 6% denaturing polyacrylamide gel and electrophoresed in 45 W for 4 to 5 h. Gels were visualized by silver staining (Promega, Madison, WI). Pixel intensity of the signal from each androgen receptor gene allele was quantified by MacBas V2.2 software (Fuji Photo Film Co. Ltd., Tokyo, Japan) on a phosphoimaging device. To ensure the accuracy of the assay, analyses were repeated on four polyclonal lesions, three monoclonal normal epithelia, and six monoclonal lesions, with starting from restriction enzyme treatment.
Interpretation of PCR Results
The X-chromosome inactivation pattern of each sample from individual cases was determined by comparing relative intensity of two allele signals between undigested and pre-PCR HhaI-digested DNA from the sample. Significant reduction (>70%) of one of two allele signals from HhaI-digested lesional DNA was viewed as evidence of monoclonality if relatively equal intensity of two allele signals or a disconcordant skewing pattern with lesional sample was found in digested DNA from matched normal control tissue. Polyclonality of the lesion was recorded when no apparent skewing of two allele signals was shown in digested lesional DNA. Cases in which control tissues had apparent skewing in the same pattern as lesional tissues were interpreted as inconclusive. In the cases in which different lesions were synchronously found, clonal lineage of different lesions was determined by comparison of monoclonality patterns between the lesions.
HPV Detection
HPV DNA was detected by single-tube nested PCR and dot blotting with slight modifications (15). Briefly, amplification was carried out in 50 μl 1 × PCR buffer, 1.5 mm MgCl2, 200 μm of each deoxynucleotide triphosphate, 1.25 U Taq polymerase, 12.5 pmol of each outer primer, and 25 pmol of each inner primer. PCR amplification products were subsequently hybridized with type-specific probes for HPV 16, 18, 31, 33, 35, 58, and 6/11 by dot-blotting technique. Amplification of β-globulin was used as control of false negativity.
RESULTS
Clonality of Precursors of Cervical Cancer
Clonality analyses were performed on 22 cases of multiple neoplastic lesions. Only lesions that provided enough cell numbers were microdissected. Altogether, 37 neoplastic lesions with different histologic severity were tested. The results from each individual case are presented in Table 1, and X-chromosome inactivation patterns of normal cervical tissues and clonality status of analyzed cervical neoplastic lesions are summarized in Table 2.
As expected, a majority (7 of 10) of samples from mixed normal stroma and epithelium gave polyclonal signals. The proportion of polyclonal signals was reduced when pure normal epithelium was examined (4 of 12 cases). Analyzed areas comprised approximately 500 normal basal cells. This suggests that the size of “tiles” of X-chromosome inactivation in ectocervical squamous epithelium analyzed may be on the order of approximately 500 basal cells.
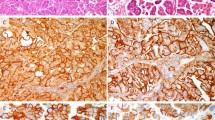
The materials were divided into two groups: 12 cases without (group A) and 10 with (group B) concomitant invasive cancer (Table 1). In addition to the inconclusive cases (2 in group A and 3 in group B) in which identical skewing X-chromosome inactivation patterns were observed in both lesional and normal control tissues, monoclonality was demonstrated in 7 cervical intraepithelial neoplasia (CIN) lesions of 15 lesions examined in group A, and all 8 tested as CIN from 7 cases in group B. Polyclonal signals were found in 5 of 15 preinvasive lesions in group A, including 1 CIN I, 2 CIN II, and 2 CIN III. In contrast, no polyclonal signals were detected in precursor lesions of group B, where there was concomitant invasive cancer. Without exception, all invasive cancers in group B scored as monoclonal. Histologic morphology and results on clonality analysis of representative cases are shown in Figure 1.

Four micrographs showing the morphology of lesions C40, C50, and C54. X-chromosome inactivation pattern was assessed by comparing electrophoretic signal intensity of the sample with (+) or without (−) HhaI digestion. Case C54 had a polyclonal signal in normal epithelium (n−; n+) and had a monoclonal one in cervical intraepithelial neoplasia (CIN) II (E−; E+) (A). C40 had equal signal intensity of both alleles in normal tissue (N−; N+) and significant reduction of the long allele CIN III (F−; F+) (B). Case C50 showed polyclonal signal in normal tissue (N−; N+), in CIN II (E−; E+) (C), and in CIN III (F−; F+) (D) (original magnification, ×200).
Genetic Relations Between Synchronous Precursors and Invasive Cancer
Ten cases of single or multiple CINs as well as synchronous invasive cancers were selected in an attempt to address the genetic links of CIN lesions to invasive cancer. Different lesions from individual cases showed no topographic continuation from each other. In 6 cases (C04, C26, C28, C76, C82, and C89), different lesions were microdissected from different tissue blocks. In four other cases (C27, C58, C83, and C88), multiple lesions were present in the same tissue slide but no direct contact between the lesions was found in three cases in addition to C83. Case C83 was the only case that showed contiguity between CIN II and CIN III but with sharp demarcation (Fig. 2).

Schematic representation of microdissections from Case C83. A, a micrograph from the border between carcinoma in situ (CIN III) and moderate dysplasia (CIN II). As seen from Table 1 and Figure 3, samples from these two anatomically adjacent precursors showed different monoclonal X-chromosome inactivation patterns. B, the invasive cancer, which showed the same X-chromosome inactivation pattern as the overlying carcinoma in situ (cf. Fig. 3) (original magnification, ×100).
Comparing X-chromosome inactivation patterns between analyzed lesions and normal control tissues, clonal relationships between CINs and synchronous invasive cancers could be determined in 7 of 10 cases. Findings compatible with a common genetic lineage between at least one preinvasive lesion and the corresponding invasive cancers could be demonstrated in five cases (C04, C27, C82, C83, C88). By contrast, discordant monoclonal patterns between precursor and invasive cancer were observed in C26, C76, and C83. In C26 and C76, clonality signals differed between CIN III and concomitant invasive cancers microdissected from different tissue blocks. In C83, the clonality signals were identical between carcinoma in situ and invasive carcinoma, whereas the CIN II had the opposite monoclonal signal. Representative electrophoretic results are depicted in Figure 3.

Results of clonal relationships between multiple synchronous neoplastic lesions of the cervix in five cases. Clonality analyses were performed as indicated in “Materials and Methods” section. Identical X-chromosome inactivation patterns between invasive cancer and coexisted cervical intraepithelial neoplasia (CIN) lesions were found in C27, C82, and C83, whereas different patterns were observed in C26, C76, and C83. N, normal tissue; n, normal epithelium; E, CIN II; F, CIN III; G, carcinoma in situ; T, invasive cancer; − and +, without and with HhaI-digestion before polymerase chain reaction amplification.
HPV Detection
As seen in Table 1, HPV was found in all invasive cancers from which samples were performed for HPV test. HPV 16 was found in six cases. HPV 18 was isolated in one case, and unidentified HPV was isolated in another case.
HPV was detected in 6 of 10 cases of group A, including HPV 16 in 1, HPV 58 in 3, and HPV 31 or 33 in the remaining 2 cases. In four monoclonal precursors, our probes did not identify HPV. Different HPV-positive lesions always carried the same type in the individual patient. When HPV was detected in normal tissues (eight cases, see description in Table 1), it was of the same type as in the respective lesions. In six cases, normal tissue was HPV negative and lesions to a variable extent were HPV positive.
DISCUSSION
Our results give a picture of the natural history of cervical cancer, which stands in contrast to that claimed for other common cancers by the same X-chromosome inactivation technique. A host of investigators have found that normal tissue will present a polyclonal X-chromosome inactivation pattern, whereas precursors such as colon polyps (5), actinic keratoses/in situ cancer of the skin (16), and corresponding invasive cancers have been monoclonal (17). This is the principal foundation for the current dogma that human cancers and benign neoplasias generally originate from a single transformed cell. Indirectly this is supported by the genetic multi-hit theory of cancer. A collection of several mutations necessary for malignant transformation is not likely to occur in many cells simultaneously (18, 19, 20)
One previous study concerning cervical cancer also largely confirmed current dogmas about cancer by showing monoclonality in invasive cancer and 30 of 30 CIN III (21). Only a single case of CIN II indicated polyclonality. The discrepancy between our results and those of that study with respect to CIN III is probably explained by our selection of cases with multiple precursor lesions in the absence of invasive cancer (group A, Table 1). Such a subgroup may either be biologically different from cases with concomitant invasive cancer (where we also always recorded monoclonality in precursors) or have been captured at such an early stage that monoclonality had not yet developed.
The long-term fate of polyclonal precursors is unknown. We could not from morphology differentiate between polyclonal and monoclonal precursors, but there was a suggestion that the type of HPV that participates in the formation of this subgroup of precursors may be unusual because they either were negative or carried the unusual HPV 33. However, only three lesions were probed for virus, and this suggestion therefore rests on uncertain ground.
Our current study observed monoclonal signals in approximately two thirds of precancers from the cases without coexisting cancers and in all precancers from the cases with accompanying cancers. Because of statistical and biologic pitfalls associated with the interpretation of monoclonality (22), we do not conclude that these prove a single-cell origin of any individual precursor of the current series. The likelihood that some have arisen from more than one cell is significant, but the fact that every invasive cervical cancer, in contrast to all other examined categories of neoplastic lesions, showed a monoclonal pattern indicates that a proportion if not all of them were indeed of monoclonal origin. To prove such a fact, it will be necessary to follow a founding mutation in multiple small microdissected samples from the cancers (23, 24).
Polyclonal signals, conversely, are not as open to biologic misinterpretation as monoclonal ones (22). Our material contains a significant number of such an outcome in a variety of precursors of invasive cancer. Because monoclonal signals may also arise from polyclonal lesions, our figures for polyclonality are minimal numbers. In all likelihood, a substantial proportion of precursors in the human cervix has therefore originated from more than one cell.
The transformation zone, because of its small size, cannot possess a large number of stem cells that are capable of undergoing neoplastic transformation. Females have a high incidence of cervical precancer. Taken together, these two facts strongly indicate an extraordinarily high risk per target cell of the transformation zone to become neoplastic. Other organs with comparable population incidence of precancer, such as colorectum, skin, or breast (25), most probably carry a much larger number of transformable cells with consequent reduction of risk per individual cell. A virus with capacity to induce multiple mutations simultaneously in many cells can be expected to explain commonly occurring polyclonality. Our results therefore indirectly support an essential role of HPV in the pathogenesis of cervical neoplasia.
Precancer of the cervix commonly regresses or at least does not regularly progress to invasive cancer (26, 27, 28). Polyclonality of precursors was observed only in the group without concomitant invasive cancers. A speculation would be that these polyclonal lesions either are predestined to regress or are caught long before any selection of invasive clones has taken place.
It is interesting that three cases were observed in which monoclonality with a different inactivated X-chromosome existed when a precursor was compared with its accompanying invasive cancer. This finding violates the concept of a direct lineage from dysplasia via cancer in situ to invasive cancer. It suggests that different neoplastic lesions may arise independently. A similar case has been published (21). It will be of value to see whether these are caused by different subtypes of HPV or an identical subtype or even variant is capable of producing varied morphologies in the same patient. In our three cases, HPV 16 was present in precursor and invasive cancer despite a different X-chromosome inactivation pattern.
Monoclonal signals from normal cervical tissue have not been reported (21). In normal epithelium, they suggest that the mosaic of X-chromosome inactivation is crude and that the tiles of the mosaic may be of uneven size and contain many stem cells that are capable of transformation. Data on the distribution of areas of X-chromosome inactivation are scanty in the human, and it is not known whether the apparent pattern in the ectocervix is common. Extensive data from mice show that mingling of cells and final sizes of tiles of identical X-chromosome inactivation vary between organs and tissues (29). Because we have not been able to analyze tile size of putative proliferative units of the transformation zone, uncertainty remains about the significance of findings in ectocervical squamous epithelium.
HPV prevalence in the group of precancers without concomitant invasive cancer in our current study deviated from the common findings in many previous studies. The reasons possibly are our selection of the cases with multiple CIN lesions and the limited number of probes applied for HPV detection. Because a limited number of cases (12 cases) were tested, this result only has a minimal value in representing the natural prevalence of HPV infection.
References
Fialkow PJ . Clonal origin of human tumors. Biochim Biophys Acta 1976; 458: 283–321.
Wainscoat JS, Fey MF . Assessment of clonality in human tumors: a review. Cancer Res 1990; 50: 1355–60.
Alexander P . Do cancers arise from a single transformed cell or is monoclonality of tumours a late event in carcinogenesis? Br J Cancer 1985; 51: 453–7.
Sternlicht M, Mirell C, Safarians S, Barsky SH . A novel strategy for the investigation of clonality in precancerous disease states and early stages of tumor progression. Biochem Biophys Res Commun 1994; 199: 511–8.
Fearon ER, Hamilton SR, Vogelstein B . Clonal analysis of human colorectal tumors. Science 1987; 238: 193–7.
Mutter GL, Chaponot ML, Fletcher JA . A polymerase chain reaction assay for non-random X chromosome inactivation identifies monoclonal endometrial cancers and precancers. Am J Pathol 1995; 146: 501–8.
Gale RE, Wheadon H, Boulos P, Linch DC . Tissue specificity of X-chromosome inactivation patterns. Blood 1994; 83: 2899–905.
Gartler SM, Riggs AD . Mammalian X-chromosome inactivation. Ann Rev Genet 1983; 17: 155–90.
Mitra AB, Murty VVVS, Li RG, Pratap M, Luthra UK, Chaganti RSK . Allelotype analysis of cervical carcinoma. Cancer Res 1994; 54: 4481–7.
zur Hausen H . Human papillomaviruses in the pathogenesis of anogenital cancer. Virology 1991; 184 (1): 9–13.
Gilliland DG, Blanchard KL, Levy J, Perrin S, Bunn HF . Clonality in myeloproliferative disorders: analysis by means of the polymerase chain reaction. Proc Natl Acad Sci U S A 1991; 88: 6848–52.
Mashal RD, Lester SC, Sklar J . Clonal analysis by study of X chromosome inactivation in formalin-fixed paraffin-embedded tissue. Cancer Res 1993; 63: 4676–9.
Guo Z, Thunberg U, Sällström J, Wilander E, Pontén J . Clonality analysis of cervical cancer on microdissected archival materials by PCR-based X-chromosome inactivation approach. Int J Oncol 1998; 12: 1327–32.
Allen RC, Zogbi HY, Moseley AB, Rosenblatt HM, Belmont JW . Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; 51: 1229–39.
Ylitalo N, Bergström T, Gyllenssten U . Detection of genital human papillomavirus by single-tube nested PCR and type-specific oligonucleotide hybridization. J Clin Microbiol 1995; 33: 1822–8.
Ren Z-P, Hedrum A, Pontén F . Human epidermal cancer and accompanying precursors have identical p53 mutations different from p53 mutations in adjacent areas of clonally expanded non-neoplastic keratinocytes. Oncogene 1996; 12: 765–73.
Williams GT, Wynford-Thomas D . How may clonality be assessed in human tumors? Histopathology 1994; 24: 287–92.
Loeb LA . Many mutations in cancer. In: Lindahl T, editor. Genetic instability in cancer. New York:Cold Spring Harbor Laboratory Press; 1996. p. 329–42.
Sidransky D, Frost P, von Eschenbach A, Oyasu R, Preisinger AC, Vogelstein B . Clonal origin of bladder cancer. N Engl J Med 1992; 326: 737–40.
Vogelstein B, Fearon ER, Hamilton SR . Genetic alterations during colorectal tumor development. N Engl J Med 1988; 319: 525–32.
Enomoto T, Haba T, Fujita M, Hamada T, Yoshino K, Nakashima R, et al. Clonal analysis of high-grade squamous intra-epithelial lesions of the uterine cervix. Int J Cancer 1997; 73 (3): 339–44.
Pontén J, Guo Z . Precancer of the human cervix. In: Pontén J, editor. Precancer: biology, importance and possible prevention. New York:Cold Spring Harbor Laboratory Press; 1998. p. 201–29.
Pontén F, Berg C, Ahmadian A . Molecular pathology in basal cell cancer with p53 as a genetic marker. Oncogene 1997; 15: 1059–67.
Pontén F, Williams C, Ling G . Genomic analysis of single cells from human basal cell using laser-assisted capture microscopy. Mutation Res 1997; 382: 45–55.
Noguchi S, Motomura K, Inaji H, Imaoka S, Koyama H . Clonal analysis of predominantly intraductal carcinoma and precancerous lesions of the breast by means of polymerase chain reaction. Cancer Res 1994; 54: 1849–53.
Gustafsson L, Adami H-O . Natural history of cervical neoplasia: consistent results obtained by an identification technique. Br J Cancer 1989; 60: 132–41.
Kottmeier HL . Evolution et traitement des épithéliomas. Rev Francais Gynécol 1955; 56: 821–5.
Nasiell K, Nasiell M, Vaclavinkova V . Behavior of moderate cervical dysplasia during long-term follow-up. Obstet Gynecol 1983; 61 (5): 609–14.
Lyon MF . X-chromosome inactivation and developmental patterns in mammals. Biol Rev 1972; 47: 1–35.
Acknowledgements
The authors thank Dr. Torsten Wahlström, Department of Gynecological Pathology, University Hospital, Helsinki, Finland, for providing some of the cases in this study.
The study was supported by grants from the National Institutes of Health (No 1 RO:CA61197–01A3) and the Swedish Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Guo, Z., Pontén, F., Wilander, E. et al. Clonality of Precursors of Cervical Cancer and Their Genetical Links to Invasive Cancer. Mod Pathol 13, 606–613 (2000). https://doi.org/10.1038/modpathol.3880104
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880104
Keywords
This article is cited by
-
Intra-tumour genetic heterogeneity and poor chemoradiotherapy response in cervical cancer
British Journal of Cancer (2011)
-
Analysis of Intratumoral Heterogeneity of Chromosome 3p Deletions and Genetic Evidence of Polyclonal Origin of Cervical Squamous Carcinoma
Modern Pathology (2001)
