
Abstract
Colorectal adenomas occur at younger age, at increased frequency and have a greater tendency for malignant transformation in patients with hereditary nonpolyposis colorectal cancer (HNPCC). We performed immunostaining for the mismatch repair proteins MLH1, PMS2, MSH2 and MSH6 in 35 colorectal adenomas from 26 patients with HNPCC and identified loss of immunostaining in 23/35 (0.66) adenomas. Loss of mismatch repair protein immunostaining was particularly frequent in large (>5 mm) (14/16) and proximally located (13/15) adenomas, whereas the gene mutated—MLH1 or MSH2—and the type of mutation did not seem to affect the results. We conclude that loss of mismatch repair protein immunostaining is detected at a lower rate in adenomas than in carcinomas associated with HNPCC. Adenomatous tissue can thus be used for immunostaining of mismatch repair proteins in clinical investigations of HNPCC, but whereas loss of immunostaining may pinpoint the gene affected and thereby guide mutation analysis, retained staining cannot exclude that the adenoma developed as part of the syndrome due to reduced sensitivity. However, the analysis has a greater chance of being informative if large and proximally located adenomas are selected.
Similar content being viewed by others

Main
Patients affected by hereditary nonpolyposis colorectal cancer (HNPCC) are at increased risk for several tumor types with the greatest risks for colorectal cancer (80% life-time risk) and endometrial cancer (40–60% risk for females). Control programmes for HNPCC-patients include colonoscopies, which aim at early identification and removal of precursor lesions. Adenomas are not numerous in HNPCC-individuals but occur at a higher frequency than in the general population. The adenomas also occur at younger age, have a predilection for the proximal colon, are larger and do more often display high-grade dysplasia and villous components.1, 2, 3 The hallmark of HNPCC-associated tumors—defective mismatch repair—is in the tumor tissue reflected as microsatellite instability and loss of immunostaining for the mismatch repair protein affected.4, 5 Mismatch repair defects are found in about 15% of sporadic colorectal cancer and in >90% of HNPCC-associated cancers.6, 7 Whereas the mismatch repair defect is generally acquired at later stages in sporadic colorectal tumorigenesis, it has been described to occur at an early stage in HNPCC tumorigenesis. Consequently, a microsatellite instability-high phenotype has been reported in 0–4% of sporadic adenomas compared to 15–20% of sporadic carcinomas.8, 9, 10, 11 Whereas the mismatch repair defects in sporadic colorectal tumors are mainly associated with somatic hypermethylation of the MLH1 promoter, HNPCC tumors develop as a result of germline mutations in these genes.7, 12
Data on the frequency of microsatellite instability in HNPCC-associated adenomas are still quite scarce, but indicate that defective mismatch repair can be found in 24–93% of the adenomas.3, 13, 14, 15, 16, 17, 18, 19 Detection of defective mismatch repair through analysis of microsatellite instability and/or loss of mismatch repair protein immunostaining in colorectal carcinomas predicts HNPCC with high sensitivity and specificity.4, 5, 20 Clinically, analysis of defective mismatch repair in an HNPCC-associated adenoma is sometimes requested since this may be the only tumor tissue available and to pinpoint the gene that mutation analysis should focus on. We therefore assessed immunohistochemical expression of the mismatch repair proteins MLH1, PMS2, MSH2, and MSH6 in 35 adenomas from 26 HNPCC patients.
Materials and methods
Patients
Among the 26 patients, 23 carried disease-causing mismatch repair gene mutations; eight different mutations in MLH1 (five of which were missense mutations) and two mutations in MSH2 (both of which were truncating). The remaining three patients (adenomas SA30, S1E and SA6) had family histories suggestive of HNPCC and loss of immunostaining for MSH2 in the tumor tissue, but no mutation had yet been identified (Table 1). Regarding type of germline mutation, 20 adenomas were derived from patients with truncating mutations or intragenic deletions and 12 from patients carrying missense mutations. Of the 35 adenomas, 30 were detected at biannual colonoscopies (although some of the larger tumors were detected at the first control) and five adenomas were found in surgical resections performed because of synchronous colorectal cancer. Adenoma size was 2–25 mm, and for five of the adenomas size was not available since they consisted of only a few adenomatous gland formations. Of the 35 adenomas, 15 were proximal, that is, located proximal to the splenic colon flexure, and 20 were distal. Multiple (2–3) adenomas from the same patients were analyzed in seven cases and these adenomas are labelled A–C (Table 1).
Immunohistochemistry
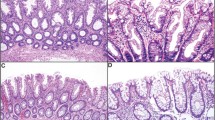
Immunostaining was performed on fresh 4-μm sections of formalin-fixed, paraffin-embedded tissue blocks and mounted on DakoCytomation ChemMate Capillary Gap Microscope slides (DakoCytomation A/S BioTek Solutions, Mt Laurel, NJ, USA) and dried at room temperature overnight followed by incubation at 60°C for 1–2 h. The tissue sections were deparaffinized in xylol and rehydrated through descending concentrations of ethanol. Antigen retrieval was achieved by microwave treatment in 1 mM EDTA, pH 9.0, at 800 W for 8 min followed by 15 min at 300 W. The slides were then allowed to cool for at least 20 min in the EDTA solution. IHC staining was performed in an automated immunostainer (TechMate 500 Plus, DakoCytomation), according to the manufacturer's instructions. The antibodies and dilutions were as follows: mouse monoclonal IgG antibodies to MLH1 (clone G168-15, dilution 1:100; PharMingen, San Diego, CA, USA), MSH2 (clone FE-11, dilution 1:100; Oncogene research products, Boston, MA, USA), MSH6 (clone 44, dilution 1:1000; BD Transduction Laboratories, Lexington, KY, USA), and PMS2 (clone A16-4, dilution 1:500, BD PharMingen). For MLH1 and MSH2 we used the detection kit LSAB™ (labelled streptavidin biotin) (DakoCytomation). MSH6 and PMS2 were stained with the EnVision™ Detection kit (DakoCytomation) with an extra enhancing step. After incubation with the primary antibody Rabbit anti-mouse immunoglobulins (DakoCytomation, dilution 1:400) were applied to the sections and they were incubated for 20 min. All tissue sections were counterstained with hematoxylin for 1 min, rinsed in running tap water for 10 min, dehydrated in ascending concentrations of alcohol and mounted. Evaluation was performed independently by two of the authors (MN and BH). A positive staining was defined as an unequivocal nuclear staining in the neoplastic cells, and nuclear staining was required in the stromal components of the tumor. A tumor cell population (which in all cases included all cells in the adenomatous components of the specimens) without nuclear staining in the presence of staining in normal epithelial, stromal, or inflammatory cells or within infiltrating lymphoid cells were classified as having lost the expression (Figures 1 and 2).

Loss of immunostaining in adenomas from HNPCC-patients with disease-causing mutations. Top: Loss of MLH1 and PMS2 staining in adenoma L2C from a patient with a nonsense mutation in MLH1. Bottom: Loss of MSH2 and MSH6 immunostaining in adenoma S1E from a patient with a frameshift mutation in MSH2.

Discordant staining patterns in two adenomas from a patient with del of exon 16 in MLH1. Adenoma SA24A shows retained staining for MLH1 and PMS2, whereas adenoma SA24B shows loss of immunostaining for MLH1 and PMS2.
Results
Loss of mismatch repair protein immunostaining was detected in 23/35 (0.66) adenomas and affected MLH1 in 14 tumors, PMS2 in 14, MSH2 in nine, and MSH6 in nine tumors (Figure 1, Table 1). Different results were found in the different subgroups. Among adenomas larger than 5 mm, 14/16 (0.88) showed loss of expression compared to 9/19 (0.47) of the adenomas 5 mm or less. Also tumor location correlated with loss of immunostaining; 13/15 (0.87) proximal adenomas showed loss compared to 10/20 (0.5) distal adenomas. In the biannual nonbaseline colonoscopy surveillance group 15 of 25 (0.6) adenomas showed loss of mismatch repair protein immunostaining, compared to 8/10 (0.8) of the remaining adenomas, but the mean adenoma size in the former group was 5 mm compared to 12 mm in the latter group. Considering the types of mutations, there was no major difference between mutations in MLH1 (loss in 14/23, 0.61) and MSH2 (loss in 9/12, 0.75) or according to type of mutation with loss in 7/12 (0.58) adenomas with missense mutations compared 12/20 (0.6) truncating mutations or intragenic deletions. We assessed microsattelite instability status (using the markers BAT25, BAT26, BAT40, BAT34, D5S346, and D2S123) in 16 of the 17 adenomas larger than 5 mm, and found MSI in 14/16 (0.88) of these adenomas. The findings of microsatellite instability did in all tumors correspond to the immunohistochemical expression pattern; that is, all 14 adenomas with loss of staining showed microsatellite instability and the two adenomas with retained mismatch repair protein immunostaining showed a microsatellite stable phenotype (Table 1). Discordant immunostaining patterns in colorectal adenomas from the same patient (Figure 2) were found in four patients (cases SA24, SA32, C1, and L2) and affected MLH1 and MSH2 in two cases each (Table 1).
Discussion
Defective mismatch repair has been described in a wide range (24–93%) of HNPCC-associated adenomas, but because of a very low (0–4%) frequency in sporadic adenomas and high frequencies of germline mismatch repair gene mutations among patients with microsatellite instable adenomas, such defects in adenomas have been suggested to predict HNPCC with high accuracy.10 Since adenomatous tissue must sometimes be used for assessment of mismatch repair defects in HNPCC investigations, we aimed at determining the frequency of loss of immunostaining for the mismatch repair proteins in adenomas from HNPCC-patients. Overall, loss of immunostaining was detected in 23/35 adenomas from 26 HNPCC-patients. All adenomas showing loss of mismatch repair protein immunostaining showed a concomitant loss of either MLH1/PMS2 or of MSH2/MSH6 (Table 1) and the analysis of microsatellite instability performed did in all cases correlate with the mismatch repair protein staining (Table 1). The overall rate of loss of immunostaining in our study, 0.66, is similar to that found by Rijcken et al,3 who demonstrated loss of immunostaining for mismatch repair proteins in 15/25 (0.6) HNPCC-associated microsatellite instability-high adenomas. We found the highest rates of loss of immunostaining in large (>5 mm) and proximal adenomas, in which the fractions of immunohistochemical loss of staining for the mismatch repair proteins were 0.88 and 0.87. Indeed, among the adenomas which did not reveal loss of immunostaining, 10/12 were located in the recto-sigmoid part of the colon and the only proximal adenoma with retained expression developed at high age and may therefore represent a sporadic lesion (Table 1).
A correlation between high-grade dysplasia and mismatch repair defects have been described.3, 18 In our series, all 13 adenomas with retained expression in our study showed low-grade dysplasia, and the four adenomas with high-grade dysplasia showed loss of staining (Table 1). In the series by Rijcken et al,3 the 10 HNPCC-adenomas with retained expression were small and had low-grade dysplasia. The observation that loss of immunostaining for the mismatch repair proteins occurs at higher rates in larger and highly dysplastic adenomas may either reflect a growth advantage inferred by the defective mismatch repair or may suggest that inactivation of the mismatch repair system occurs during tumor progression rather initiation. If the adenomas defective in mismatch repair have a growth advantage, this may explain why these tumors are larger and show frequent high-grade dysplasia, and we did indeed identify loss of immunohistochemical mismatch repair protein staining in several adenomas that were only a few millimeters in size. Alternatively, tumor initiation could depend on, for example, a mutation in the APC gene, whereafter the defective mismatch repair accelerates tumor progression, presumably through an accumulation of somatic frameshift alterations in various cancer-associated genes.1 Although studies of molecular alterations in adenomas have not identified any major differences between sporadic and HNPCC-associated adenomas, and if also the HNPCC-associated adenomas achieve the mismatch repair-defective phenotype through somatic hypermethylation of the MLH1 promoter, this could contribute to the relative predominance of proximal carcinomas in HNPCC.21
Hyperplastic polyps are generally small (they rarely exceed 1 cm in size), commonly occur within the large bowel, and have traditionally been viewed as non-neoplastic. However, clonal genetic changes—including mutations of KRAS and BRAF and microsatellite instability—have been demonstrated in hyperplastic polyps.11, 15, 18, 19, 22, 23, 24 These lesions have been associated with an increased risk of malignancy in the hyperplastic polyposis syndrome, but may signify an increased risk of colorectal cancer also in other individuals.18, 22 In HNPCC as well as in familial colorectal cancer, a strong correlation has been found between adenomas and hyperplastic polyps, suggesting similar mechanisms for their initiation.25, 26 We investigated 12 hyperplastic polyps that had occurred in the 26 patients in the study and found retained immunostaining for all four mismatch repair proteins in all polyps (data not shown), which is in accordance with previous studies and suggests that the value of immunostaining in these lesions is of limited clinical value.27 Serrated adenomas have been suggested to represent more specific precursors of microsatellite instable tumor development and show distinct molecular features, such as microsatellite instability and a low frequency of KRAS mutations, but the serrated pathway is also heterogenous.28 Only one serrated adenoma was included in our series and this tumor occurred in conjunction with an MSH2 mutation and showed loss of MSH2/MSH6 protein expression.
In conclusion, immunostaining for defective mismatch repair proteins in HNPCC-associated adenoma has a high specificity and can thereby direct the mutation screening to the gene affected. However, loss of immunostaining for these proteins occurs at a lower frequency in adenomas than in carcinomas, and depends on the size and location of adenoma. From a clinical diagnostic point of view, retained immunostaining for the mismatch repair proteins in small (<5 mm) and left-sided adenomas cannot be used to exclude HNPCC, whereas loss of staining in a patient suspected of having HNPCC may be used to direct mutation analysis.
References
Jass JR, Stewart SM, Stewart J, et al. Hereditary non-polyposis colorectal cancer—morphologies, genes and mutations. Mutat Res 1994;310:125–133.
Lindgren G, Liljegren A, Jaramillo E, et al. Adenoma prevalence and cancer risk in familial non-polyposis colorectal cancer. Gut 2002;50:228–234.
Rijcken FEM, Hollema H, Kleibeuker JH . Proximal adenomas in hereditary non-polyposis colorectal cancer are prone to rapid malignant transformation. Gut 2002;50:382–386.
Lindor NM, Burgart LJ, Leontovich O, et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol 2002;20:1043–1048.
Halvarsson B, Lindblom A, Rambech E, et al. Microsatellite instability and/or immunostaining for the diagnosis of hereditary nonpolyposis colorectal cancer? Virchows Arch 2004;444:135–141.
Thibodeau SN, Bren G, Schaid D . Microsatellite instability in cancer of the proximal colon. Science 1993;260:816–819.
Mitchell RJ, Farrington SM, Dunlop MG, et al. Mismatch repair genes hMLH1 and hMSH2 and colorectal cancer: a HuGE review. Am J Epidemiol 2002;156:885–902.
Young J, Leggett B, Gustafson C, et al. Genomic instability occurs in colorectal carcinomas but not in adenomas. Hum Mutat 1993;2:351–354.
Samowitz WS, Slattery ML . Transforming growth factor-B receptor type 2 mutations and microstatellite instability in sporadic colorectal adenomas and carcinomas. Am J Pathol 1997;151:33–35.
Loukola A, Salovaara R, Kristo P, et al. Microsatellite instability in adenomas as a marker for hereditary nonpolyposis colorectal cancer. Am J Pathol 1999;155:1849–1853.
Konishi K, Yamochi T, Makino R, et al. Molecular differences between sporadic serrated and conventional colorectal adenomas. Clin Cancer Res 2004;10:3082–3090.
Cunningham JM, Christensen ER, Tester DJ, et al. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res 1998;58:3455–3460.
Aaltonen LA, Peltomäki P, Mecklin JP, et al. Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res 1994;54:1645–1648.
Jacoby RF, Marshall DJ, Kailas S, et al. Genetic instability associated with adenomas to carcinoma progression in hereditary nonpolyposis colon cancer. Gastroenterology 1995;109:73–82.
Konishi M, Kikuchi-Yanoshita R, Tanaka K, et al. Molecular nature of colon tumor in hereditary nonpolyposis colorectal cancer, familial polyposis and sporadic colon cancer. Gastroenterology 1996;111:307–317.
Akiyama Y, Iwanaga R, Saitoh K, et al. Transforming growth factor B type II receptor gene mutations in adenomas from hereditary nonpolyposis colorectal cancer. Gastroenterology 1997;112:33–39.
Sasaki S, Masaki T, Umetani N, et al. Microsatellite instability is associated with the macroscopic configuration of neoplasms in patients with multiple colorectal adenomas. Jpn J Clin Oncol 1998;28:427–430.
Iino H, Jass JR, Simms LA, et al. DNA microsatellite instability in hyperplastic polyps, serrated adenoma, and mixed polyps: a mild mutator pathway for colorectal cancer? J Clin Pathol 1999;52:455–460.
Iino H, Simms L, Young J, et al. DNA microsatellite instability and mismatch repair protein loss in adenomas presenting in hereditary non-polyposis colorectal cancer. Gut 2000;47:37–42.
Liu T, Wahlberg S, Burek E, et al. Microsatellite instability as a predictor of a mutation in a DNA mismatch repair gene in familial colorectal cancer. Genes Chromosomes Cancer 2000;27:17–25.
Voskuil DW, Kampman E, van Geloof W, et al. No major difference in K-ras and p53 abnormalities in sporadic and hereditary nonpolyposis colorectal adenomas. Drug Dis Sci 2000;45:2187–2194.
Rashid A, Houlihan PS, Booker S, et al. Phenotypic and molecular characteristics of hyperplastic polyposis. Gastroenterology 2000;119:323–332.
Chan TL, Zhao W, Leung SY, et al. Cancer Genome Project. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res 2003;63:4878–4881.
Kambara T, Simms LA, Whitehall VLJ, et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004;53:1137–1144.
Liljegren A, Lindblom A, Rotstein S, et al. Prevalence and incidence of hyperplastic polyps and adenomas in familial colorectal cancer: correlation between the two types of colon polyps. Gut 2003;52:1140–1147.
Goldstein NS, Bhanot P, Odish E, et al. Hyperplastic-like colon polyps that preceded microsatellite-unstable adenocarcinomas. Am J Clin Pathol 2003;119:778–796.
Rijcken REM, van der Sluis T, Hollema H, et al. Hyperplastic polyps in hereditary nonpolyposis cancer. Am J Gastroenterol 2003;98:2306–2311.
Sawyer EJ, Cerar A, Hanby AM, et al. Molecular characteristics of serrated adenomas of the colorectum. Gut 2003;51:200–206.
Acknowledgements
Eva Rambech and Kristina Lövgren are acknowledged for technical assistance. The study was financially supported by grants from the Swedish Cancer Society, the Nilsson Cancer Foundation, the Gustaf V Cancer Foundation, the Kamprad Cancer Foundation, and the Region Skane Research Founds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Halvarsson, B., Lindblom, A., Johansson, L. et al. Loss of mismatch repair protein immunostaining in colorectal adenomas from patients with hereditary nonpolyposis colorectal cancer. Mod Pathol 18, 1095–1101 (2005). https://doi.org/10.1038/modpathol.3800392
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800392
Keywords
This article is cited by
-
Prevalence of Mismatch Repair-Deficient Colorectal Adenoma/Polyp in Early-Onset, Advanced Cases: a Cross-Sectional Study Based on Iranian Hereditary Colorectal Cancer Registry
Journal of Gastrointestinal Cancer (2021)
-
Microsatellite instability screening in colorectal adenomas to detect Lynch syndrome patients? A systematic review and meta-analysis
European Journal of Human Genetics (2020)
-
Mismatch repair deficient-crypts in non-neoplastic colonic mucosa in Lynch syndrome: insights from an illustrative case
Familial Cancer (2015)
-
Advanced Colorectal Adenomas in Patients Under 45 Years of Age Are Mostly Sporadic
Digestive Diseases and Sciences (2014)
-
Interval colon cancer in a Lynch syndrome patient under annual colonoscopic surveillance: a case for advanced imaging techniques?
BMC Gastroenterology (2012)
